





Introduction
- Erlich put forward the term ‘horror autotoxicus’ to refer to the body’s aversion to immunological self destruction, and postulated that mechanisms must prevent reaction against self
- Healthy individuals have a smouldering autoimmune response that can escape control (a system of many checks and balances) to cause autoimmune disorders
- A major area of interest is how the immune system discriminates between self and non-self, and thus how to minimise the possibility of autoimmunity while optimising protective immune responses
- Active mechanisms also prevent an immunologic attack on harmless antigens such as those represented by commensal bacteria and food
- Control mechanisms induce a state of immune unresponsiveness to an antigen
- Tolerance is induced and maintained both centrally and peripherally, each with a non-redundant function in maintaining receptor diversity while curtailing self-reactivity
- It was observed more than 50 years ago that non-identical twin calves sharing a placenta do not react to each other’s erythrocytes, and it was hypothesised that in utero exposure rendered them tolerant to each other’s antigens
- This spurred Medawar and Brent to inject mouse cells into a neonatal mouse from a different strain, resulting in tolerance of the latter to skin grafts from the former [see their original paper, “Actively Acquired Tolerance of Foreign Cells”; downloadable at end of this section]
- MacFarlane Burnet pioneered the clonal selection theory: where host encounter with a foreign antigen selects for a particular immune cell clone that then proliferates to yield daughter clones which all have the same specificity
- He hypothesised that self-tolerance resulted from central deletion of ‘forbidden clones’, thereby eliminating potentially self-reactive clones
- additionally, a deficiency in Treg number or function leads to allergic, autoimmune and other inflammatory diseases due to loss of tolerance
- The most important form of tolerance is self tolerance, which occurs in the developing foetus during normal immune ontogeny
- Tolerance can also be induced to non-self antigens after birth
- An antigen that induces tolerance is termed a tolerogen and tolerance is both antigen- and cell clone-specific: tolerance is selective for the tolerogen that induced it, facilitating continued responsiveness to other antigens
- In this way, tolerance is different from generalised immune suppression (such as that induced by post-transplant drugs like cyclosporine)
Central vs. Peripheral Tolerance
- Induction of tolerance requires education of both B and T cells, which occurs in both central (bone marrow, thymus) and peripheral (spleen, lymph nodes) lymphoid organs and tissues
- Here lymphocytes become either immune competent or tolerant towards encountered antigens
- Mechanisms of tolerance induction and maintenance differ between B and T cells, and between central and peripheral lymphoid organs
Central T-Cell Selection
- Transgenic animal models demonstrate that central mechanisms are indispensable for induction of self-tolerance
- CD4-CD8- (double negative) T-cell progenitors enter the thymic cortex and rearrange their receptors to become CD4+CD8+ (double positive) thymocytes
- Positive and negative selection occurs in the thymus (FIGURE 1)
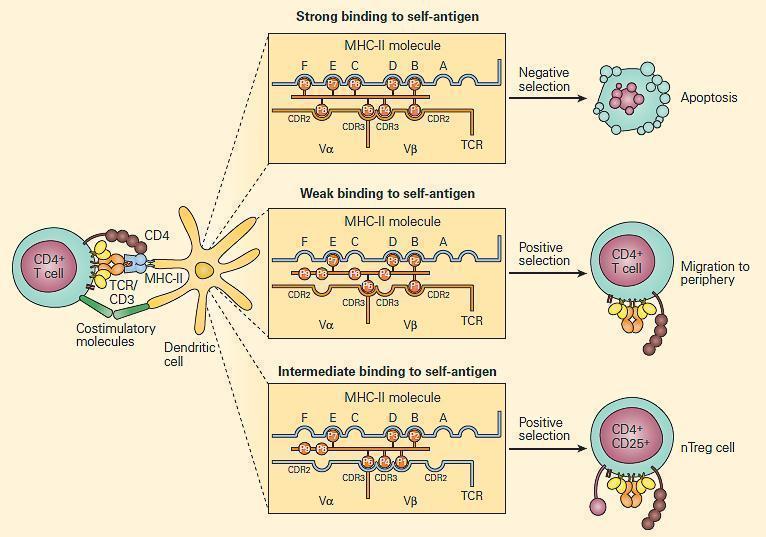
Figure 1. Positive and negative selection in the thymus. CD4+ T cells that recognize self-antigens expressed on thymocytes in the context of MHC-II molecules undergo apoptosis. The key factor in determining positive and negative selection is the strength of the antigen recognition by the maturing T cell; low-avidity recognition leads to positive selection, and high-avidity recognition induces negative selection. It is proposed that at this stage, Treg (CD4+CD25+) cells that are autoantigen-specific are generated by intermediate degrees of binding. [Reproduced with permission from Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012]
- T-cells with a receptor that bind with moderate affinity to self-peptide-MHC complexes on thymic epithelia receive a survival signal (positive selection)
- Depending on which MHC was recognised, the T-cell will display either CD4 or CD8 (single positive)
- Negative selection occurs at the DP stage in the cortex, or at the SP stage in the medulla: T-cells with a receptor that bind with high avidity to autoantigens on thymic epithelia undergo apoptosis
- The autoantigens are host tissue proteins expressed on thymic epithelia under regulation of the transcription factor autoimmune regulator (AIRE)
- Many T-cells are eliminated: of the potential 109 receptor specificities in the thymus, only a fraction are present in peripheral tissues
- AIRE deficiency results in organ-specific autoimmunity, including APS-1 (damage to parathyroid and adrenal glands)
Peripheral T-Cell Selection
- Central and Peripheral tolerance occur in tandem, in the case that central tolerance is not completely effective; partly because not all autoantigens are expressed in the thymus
- Several autoreactive clones are found in the peripheral blood of healthy people, and some lymphocytes from people without MS react in vitro to MBP (a target of the immune response in MS)
- Autoreactive clones can potentially become activated and proliferate in the periphery when properly stimulated (e.g. sub-acute bacterial endocarditis can lead to emergence of self-reactive clones that damage the kidneys)
- Peripheral mechanisms of tolerance eliminate or suppress autoreactive clones that escape to the periphery
- Mechanisms of peripehral T-cell tolerance include:
- A. Clonal deletion
- B. Ignorance
- C. Anergy
- D. Immune regulation
- Tolerance mechanisms can also result in inappropriate tolerance to non-self antigens.
- As an example: when LCM virus was inoculated into mouse embryos, adult mice mounted no immune response to LCM and were chronically infected
- It was thought that these antigens, introduced early during gestation, were handled as self-antigens, thereby inducing negative central T-cell selection.
A. Clonal Deletion
- The best-studied mechanism eliminating activated T-cell clones is activation-induced cell death (FIGURE 2)
- T-cells activated by antigen-presenting cells express IL-2 and IL-2R for autocrine facilitation of proliferation
- Activated T-cells also increase their expression of death receptors (e.g. Fas) and their ligands
- Ligation of Fas leads to T-cell apoptosis via the caspase pathway, thereby ending the immune response
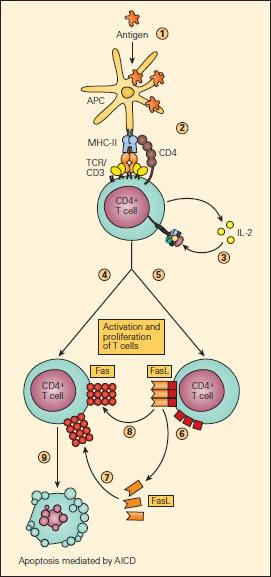
Figure 2. Schematic representation of AICD. Upon uptake and processing of antigen by APCs (1) and subsequent presentation of the processed peptide to a CD4+ T cell (2), IL-2 production and expression of the IL-2R occurs followed by their autocrine binding (3), leading to T cell activation. Activated T cells express Fas (4) and FasL (5) on their surfaces or as soluble s-FasL after cleavage of the membrane-associated FasL (6). The interaction of Fas either with s-FasL (7) or with membrane-associated FasL (8) leads to apoptosis (9) by activation-induced cell death (AICD), thus ending the immune response [Reproduced with permission from Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Mutations in Clonal Deletion Pathways
- Mice whose T-cells do not express Fas, display expanded lymphocyte populations in their secondary lymphoid organs, leading to profound lymphadenopathy, significant self-reactivity (including many autoantibodies), and autoimmune damage to many organs (including kidneys)
- Rare human diseases, ALPS 1a and 1b, are caused by similar mutations in Fas or FasL respectively, also leading to lymphadenopathy and autoantibodies
- IL-2 affects the Fas pathway, and can eventually lead to AICD by increasing FasL expression
- IL-2 also helps attenuate immunity by downregulating survival molecules (e.g. FLICE and FLIP) which would otherwise inhibit AICD by preventing assembly of the Fas death receptor complex
B. Ignorance
- Although peripherally, T-cells from healthy individuals can react with self-antigens in vitro, this does not commonly occur in vivo
- It is thought that T-cells ignore certain self-antigens because they are located in immune-privileged sites or because they have low immunogencitiy (low levels of expression or low binding affinity)
- Sympathetic autoimmune ophthalmia (Case 38 in Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012 and related Case Study – Case of eye injury and decreased vision) – severe inflammatory damage to both eyes – is caused by release of sequestered ocular self-antigens into circulation, where they can eventually activate peripheral autoreactive immune cells
- the immune system is not normally exposed to ocular antigens, but trauma to a single eye releases autoantigens that activate autoreactive immune cells, leading to severe granulomatous inflammatory of both eyes
C. Anergy
- This is a major mechanism inactivating peripheral autoreactive T-cell clones
- Anergic T-cell clones cannot respond to cognate antigenic stimuli: they do not produce IL-2 or IL-2R
- Multiple proposed mechanisms explain this block in T-cell activation (FIGURE 3):
- Disruption of the interaction between the T-cell co-receptor CD28 and APC co-stimualtory molecules CD80/86
- Interaction of CTLA-4 with CD80/86, negatively regulating T-cell activation
- T-cells displaying CD28 tend to be activated by APC, while T-cells displaying CTLA-4 tend to become anergic
- Under physiologic conditions, T-cells express CD28 on initial encounter with APC
- Shortly after such stimulation, they start displaying CTLA-4, which has a higher binding affinity than CD28 for CD80/86 than does CD28
- CTLA-4 knockout mice develop profound lympho-proliferative disease
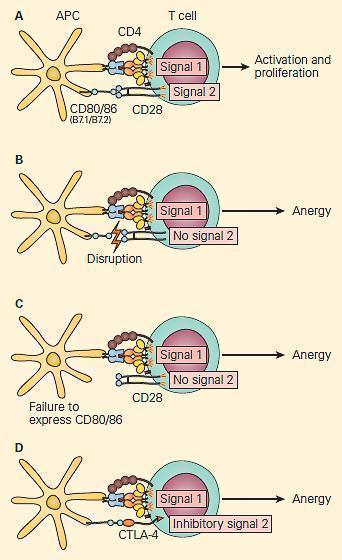
Figure 3. Co-stimulation is important for the activation of T cells. Panel A: Following the activation of the 80/86 on the antigen-presenting cell provides the second signal, leading to T cell activation and proliferation. Panel B: Disruption of the CD28/CD80/86 signal or Panel C: Failure to express CD 80/86 can lead to anergy. Panel D: At the same time, the activated T cells upregulate the expression of CTLA-4, a molecule that also interacts with greater affinity to CD80/86, leading to disruption of the costimulatory signal and anergy. [Reproduced with permission from Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012]
Clinical Significance of CTLA-4
- Rheumatoid arthritis is an autoimmune disease mainly affecting the joints
- CTLA-4 is used as a biologic response modifier to treat these patients, significantly reducing joint inflammation
- A fusion protein consisting of CTLA-4 and immunoglobulin (abatacept, belatacept) is used clinically in arthritis and after transplantation
- There is also interest in blocking CTLA-4 using monoclonal antibodies (e.g. ipilimumab) to inhibit tumor tolerance
- Insufficient production of key transcriptional activators (e.g. AP-1, NF-kB, NFAT-1) during T-cell activation reduces IL-2 production
- Transcription suppressors (e.g. CREM) can also decrease transcriptional activity of the IL-2 gene promoter, leading to T-cell anergy
- Alterations at multiple levels of regulation (e.g. activity of key kinases such as MAPK, or stability of IL-2 mRNA) can contribute to reduced IL-2 production in anergic T-cells
D. Immune Regulation
- Immune regulation is achieved by the action of Treg’s
- Treg are important in the maintenance of peripheral tolerance
- When they are depleted from mice, autoimmunity results
- A human patient with genetic Treg dysfunction develops lymphadenopathy and inflammatory infiltrates consisting of autoreactive T-cells in multiple organs
- Naturally-occurring thymus-derived Treg display anergic properties in vitro, but can also suppress CD4+CD25- T-cells in vivo, via direct cell-cell contact, or secretion of cytokines (FIGURE 4)
- During active inflammation (e.g. infection), Treg do not prevent protective immune function
- Induced CD4+CD25+ Treg can also be activated in peripheral lymphoid organs (e.g. by TGFβ) and suppress immune responses via anti-inflammatory cytokines (e.g. TGFβ) rather than direct contact.
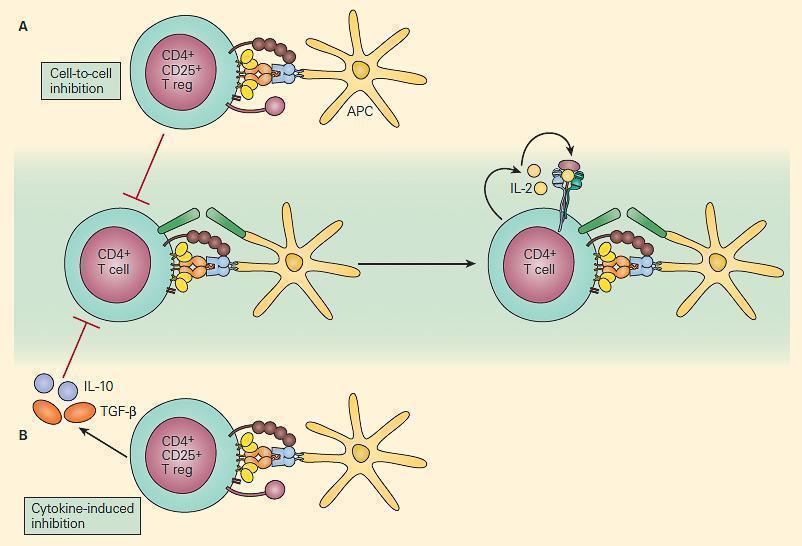
Figure 4. Schematic representation of two mechanisms of immune regulation by Treg cells. Panel A: Self-antigen specific CD4+CD25+ T cells can directly suppress the activation of self-reactive T cells through cell-cell interaction. Panel B: Anti-inflammatory cytokines such as IL-10 and TGF-β that are secreted by other immunoregulatory T cells can also suppress the immune response. [Reproduced with permission from Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012]
TGFβ & Regulatory Cells Other Than Treg
- TGFβ in the presence of IL-6 (e.g. from activated macrophages during infection) and IL-23 can also lead to induction of Th17, which produce IL-17
- Th17 are associated with antimicrobial immunity as well as autoimmune/inflammatory disorders
- TGFβ can therefore either regulate inflammation through Treg, or promote inflammation through Th17
- CD1-restricted NKT cells have also been implicated in immune regulation, as have CD8+ suppressor T-cells
- γδCD8+ T-cells that populate MALT are most likely involved in suppression of immune responses initiated by antigen delivered by the mucosal route
- After inhalation of small quantities of antigen, such CD8+ T-cells are activated in sub-mucosal areas to become suppressors cells and migrate to draining lymph nodes to suppress immune response via production of IL-10 and TGFβ
- Ingestion of larger quantities of antigen activates not only such CD8+ suppressors, but also Treg that migrate to areas of inflammation to downregulate T-driven immune responses
- Th1-type IFNγ opposes Th2-type immunity, while Th2-type IL-4 opposes Th1-type immunity
- Regulatory T-cells and cytokines are also being used as and targeted therapeutically
B-cell Tolerance
- During normal B-cell development, a set of processes help induce B-cell central tolerance
- Education of B-cells and elimination of self-reactive B-cell clones is somewhat different from that of T-cells
- B-cells are still immature when they relocate from bone marrow to spleen T-cell zones
- Autoreactive B-cells are not necessarily eliminated during negative selection in the bone marrow
- B-cells that recognise autoantigens are eliminate via apoptosis or become anergic
- Autoreactive B-cells that escape negative selection become part of the a maximally-diverse immune repertoire
B-cell Peripheral Tolerance
- Peripheral tolerance mechanisms (in secondary lymphoid tissues) exist for various reasons:
- Imperfect T-cell tolerance: in most autoimmune diseases, B-cells are T-cell dependent, requiring help from pre-activated cognate autoreactive T-cells
- T-independent B-cells can be activated by autoantigens without T-cell help
- Microbial antigens structurally similar to autoantigens can lead B-cells to produce cross-reactive antibodies in a phenomenon known as molecular mimicry
- B-cells hypermutate their receptors on activation, so there is a second chance that they may become self-reactive
Exploring B-cell Tolerance
- Mechanisms of B-cell tolerance have been explored using transgenic mice (FIGURE 5)
- In a classic experiment, monotransgenic mice expressing hen egg lysozyme (after tolerisation via fetal exposure) are bred with monotransgenic mice expressing anti-lysozyme IgM and having B-cell receptors capable of recognising lysozyme
- The resulting mouse hybrids produce lysozyme, but despite lysozyme-recognising B-cells do not produce anti-lysozyme antibody after immunization with lysozyme
- This demonstrates that the simultaneous presence of lysozyme and lysozyme-reactive B-cells leads to B-cell anergy
- B-cells from the hybrid strain transferred into irradiated wild-type mice were indeed able to make anti-lysozyme antibodies
- This proves that anergy was related to continued stimulation by lysozyme, but could be reversed when B-cells were in a host without the tolerising effects of lysozyme
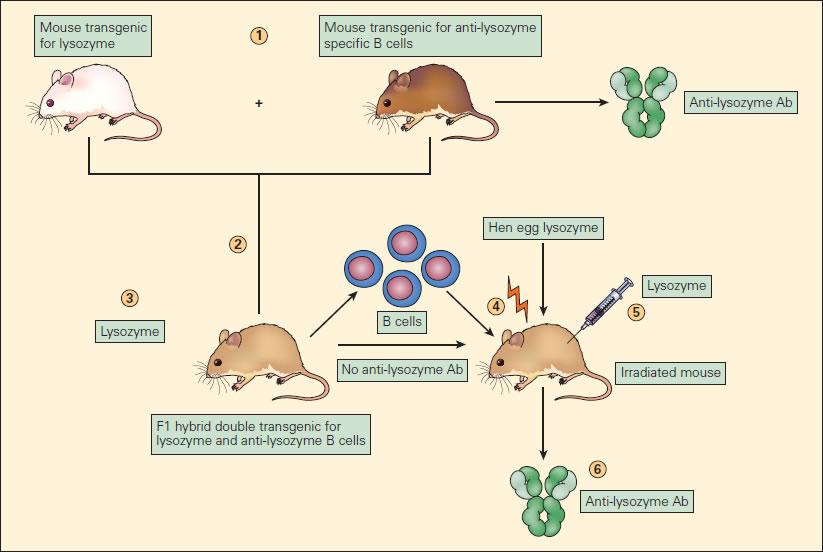
Figure 5. Experimental model of B cell anergy. (1) A transgenic mouse expressing hen egg lysozyme (white mouse) was crossed with a mouse transgenic for anti-lysozyme specific B cell production (brown mouse) to produce an F1 hybrid (tan mouse) (2) double transgenic for lysozyme production (3) and anti-lysozymal B cell production; these double transgenic hybrids produced lysozyme but did not produce anti-lysozyme antibody. (4) The B cells from these F1 mice, although unable to react to the lysozyme, when transferred to irradiated mice and immunized with lysozyme (5) resulted in the production of anti-lysozyme antibody (Ab) (6). This proves that the B cells in the F1 mice, although capable of producing anti-lysozyme antibody, if transferred to another mouse were functionally anergized in the presence of endogenously produced lysozyme [Reproduced with permission from Bellanti JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Regulatory B Cells & Tolerance
- B cells are known to be antibody producers. In fact more specifically, B stimulation occurs in the Germinal Centres of the lymph nodes and develop into antibody secreting plasma cells with the help of T follicular cells.
- This is a well described pathway and there are many reviews on the topic
- What is less well known, is the regulatory nature of B cells and how these cells can play a role in regulating immunity, additional to the antibody functions of these cells.
Regulatory B-cells
- Regulatory B-cells (Breg) are a B-cell sub-population present in mice and humans.
- They are important in maintaining immune homeostasis (e.g. they contribute to maintenance of tolerance, and prevention of uncontrolled inflammation).
- They can also secrete immunoglobulins (especially IgM), however.
- Breg origins are incompletely understood, but they may develop from marginal zone B-cells, from CD5+ transitional or pre-naïve B-cells, or from the human equivalent of B1 cells.
- Triggers for Breg development include inflammation, the presence of apoptotic cells, tolerogenic DC (tolDC)-derived IFNβ, tumor-related TNFα, and interaction with tolDC CD40 or activated helper T-cell surface CD40L. B-cell activating factor (BAFF) and T-cell surface CTLA-4 may also play roles.
- There is no unequivocal Breg identity, possibly because Breg are a transient phenotype along. The current best definition in humans is CD19+CD24hiCD38hi (equivalent to transitional B-cells), although heterogeneity exists.
- Various Breg subsets include:
- Immature transitional (CD19+CD24hiCD38hi) B-cells – the peripheral B-cell subset producing the highest levels of IL-10.
- B10 cells (mainly within the CD19+CD24hiCD27+ population) – a rare circulating sub-population related to memory B-cells and characterised by IL-10 production as well as a high proliferative capacity on activation, with fully IL-10-dependent regulatory abilities. Some have autoreactive B-cell receptors, and there is interest in elucidating their relationship to the human equivalent of B1 cells.
- GrB+ (CD19+CD38+CD1d+IgM+CD147+) B-cells – induced by T-cell-derived IL-21, and expressing IL-10, IDO, and granzyme B.
- Br1 (CD25hiCD71hiCD73lo) cells – secrete allergen-specific tolerogenic IgG4.
- Plasmablasts (CD27intCD38hi) – also capable of IL-10 secretion
- Circulating CD39+CD73+ Breg – express enzymes which convert pro-inflammatory ATP to adenosine.
- iBreg (phenotype undefined) – induced by T-cell surface CTLA-4, express TGFβ and IDO.
- Subsets down-regulate innate and adaptive responses via diverse molecular mechanisms, including secretion of regulatory cytokines (e.g. IL-10, TGFβ), expression of regulatory enzymes (e.g. IDO), and direct cell-cell contact (e.g. antigen presentation along with co-stimulation by CD80/86, CD40; or cytotoxicity via granzyme B or diverse TNF and TNF receptor (TNFR) family members, such as Fas/FasL, TRAIL/DR5, and PDL1 and -2).
- In many cases, suppression is IL-10-dependent, yet IL-10 is not always necessary for Breg suppressive function. The multitude of Breg suppressive mechanisms means that demonstration of suppressive capacity remains the gold standard for Breg
- Results of Breg suppressive mechanisms include inhibition of T-cell proliferation, suppression of Th1 and Th17 cytokine (IFNγ, TNFα, IL-17) production, restoration of the Th1/Th2 balance, promotion of T-cell IL-10 secretion, Treg induction, inhibition of dendritic cell and macrophage function (e.g. phagocytosis, antigen presentation, TNF secretion, NO production), and Breg surface CD1d may facilitate activation of iNKT cells with regulatory functions.
- Clinically Breg are implicated in inflammation, autoimmunity (e.g. animal models of arthritis, multiple sclerosis, type I diabetes, systemic lupus erythematosus(SLE)), allergy (e.g. animal models of contact hypersensitivity), cancer, and transplantation. Breg appear to play both tumor-promoting and tumor-inhibiting roles.
- Most studies on Breg have been done in vitro or in murine models; less is known about Breg functions in healthy humans and human autoimmune diseases. Although autoimmune patients display an increased Breg frequency, Breg from SLE patients have impaired suppressive activity due to a defect in IL-10 production
Autoimmunity: a breakdown of tolerance
- The immune system’s three basic functions are Defence, Surveillance and Homeostasis
- Elaborate and redundant tolerance mechanisms are in effect both during maturation and later in the lymphocyte life-cycle, leading to clonal deletion or anergy
- Anergy can be reversed to allow recruitment of autoreactive clones to maximise receptor diversity (e.g. during infection)
- Autoimmunity [Table 1] emerges when self-tolerance mechanisms fail (infections can lead to breaking of tolerance [Case Studies – A 9 year old girl presents with body swelling, shortness of breath and backache and Why can I not walk today?]
- Primary immunodeficiencies can also present with altered surveillance (leading to malignancy) or altered homeostasis (leading to autoimmunity)
- Breaking tolerance usually occurs as a consecutive series of many events (rarely due to a single genetic/environmental factor).
- Breaking of tolerance can be conceptualised as being set in motion by just the right stimuli occurring against the backdrop of a predisposing immunological milieu
- This milieu is influenced by genetics, prior antigen encounter, local factors in target organs, and other endogenous factors (e.g. the immune-modulating effects of hormones such as cortisol and oestrogen)
- Autoreactive T-cells can, for example, proliferate peripherally during an infection or on release of previously-sequestered antigens, or defects of apoptosis can facilitate development of T-cell autoreactivity
- Similar mechanisms can lead to proliferation of autoreactive B-cells
- Such events collectively lead to dysregulation and loss of tolerance
Table 1. Partial list of autoimmune diseases
| Disease | Main organ affected | Proposed self-antigen(s) | Clinical presentation |
|---|---|---|---|
| Organ-specific autoimmune diseases | |||
| Multiple sclerosis | Central nervous system | Myelin basic protein, myelin oligodendrocyte protein | Loss of vision, weakness of limbs, sensory abnormalities, incontinence |
| Sympathetic ophthalmia | Eye | Various uveal antigens | Eye pain, loss of vision, sensitivity to light |
| Graves' disease | Thyroid | Thyrotropin receptor | Hyperthyroidism (weight loss, nervousness, palpitations, diarrhea), exophthalmos |
| Hashimoto's thyroiditis | Thyroid | Thyroperoxidase, thyroglobulin | Hypothyroidism (weight gain, constipation, skin changes, myxedematous dementia) |
| Goodpasture's syndrome | Lung, kidney | Glomerular basement membrane (type IV collagen) | Kidney and respiratory insufficiency |
| Pernicious anemia | Stomach | Intrinsic factor | Anemia, gastritis |
| Crohn's disease * | Intestine | ? microbial antigens | Hemorrhagic diarrhea, abdominal pain, draining fistulas |
| Ulcerative colitis * | Large Intestine | ? microbial antigens | Hemorrhagic diarrhea, abdominal pain |
| Diabetes mellitus type I | Pancreas | Islet cell, insulin, glutamic acid decarboxylase (GAD) | Polyphagia, polyuria, polydipsia, weight loss |
| Immune thrombocytopenia | Platelets | Glycoproteins on the surface of platelets | Easy bruising, hemorrhage |
| Myasthenia gravis | Muscle | Acetylcholine receptor | Muscle weakness, fatiguability |
| Hemolytic anemia | Red cells | I antigen | Anemia |
| Systemic autoimmune diseases | |||
| Sjögren's syndrome | Salivary and lacrimal glands | Nuclear antigens (SSA, SSB) | Dry eyes, dry mouth, lung and kidney disease |
| Rheumatoid arthritis | Joints, lung, nerves | Citrulinated peptides in the joint, IgG | Deforming arthritis, skin nodules, occasional lung and nerve involvement |
| Wegener's granulomatosis | Lung, kidney | Proteinase 3 (c-ANCA) | Sinusitis, shortness of breath, kidney failure |
| Systemic lupus erythematosus | Kidney, skin, joints, central nervous system | DNA, histones, ribonucleoproteins | Arthritis, skin rashes, kidney insufficiency, nerve damage |
*Although previously considered autoimmune diseases, more recent evidence supports that they are autoinflammatory disorders
Autoimmune Response vs. Autoimmune Disease
- A clear distinction must be made between an autoimmune response and an autoimmune disease.
- The term autoimmune response refers to the demonstration of an autoantibody or T cell-mediated reactivity directed to a self-antigen.
- An autoimmune response may or may not be associated with autoimmune disease. For example, many clinically well individuals, particularly women, exhibit an autoimmune response by the presence of serum antinuclear antibodies (ANAs) and demonstrate no symptoms.
- Although the presence of ANA may also be associated with the autoimmune disease SLE, the diagnosis requires the presence of additional clinical features of the disease, e.g., rash, arthritis, or kidney involvement.
- Although much progress has been made in understanding the pathophysiology of autoimmune diseases, the underlying etiopathogenesis remains elusive for most of these disorders.
- Autoimmunity affects ~5% of the population, predominantly women in their reproductive years, with peak incidences during adolescence and the fourth through fifth decades
- An understanding of lymphocyte physiology and tolerance mechanisms largely derives from transgenic animal models, as well as genomics and proteomics research
- There has been some success translating such observations to humans, but additional clinical studies are required, including to better understand the pathogenesis of autoimmunity
- Improved knowledge may be applied to generate more efficacious and safer autoimmune therapies
Organ-specific vs. Systemic Autoimmunity
- Organ-specific autoimmunity is targeted against a single organ while systemic autoimmunity affects diverse tissues
- It is not uncommon for a patient to have symptoms of more than one autoimmune disease (known as ‘overlap’, or undifferentiated collagen vascular syndrome)
- In addition, a patient with one autoimmune disease may have serologic markers – but no clinical manifestations – of another
- An autoimmune response is defined as demonstration of autoantibodies or T-cell autoreactivity, which may or may not be associated with clinically manifested autoimmune disease
- Many healthy women have serum antinuclear antibodies, but demonstrate no symptoms of SLE (whose diagnosis requires the presence of clinical features such as rash, arthritis, and renal involvement)
Classification of T cell and Antibody mediated diseases
- Table 2 shows groups of some autoimmune diseases mediated by either T cell autoreactivity or by auto-antibodies:
| Antibody-mediated autoimmune diseases | T cell mediated autoimmune diseases |
|---|---|
| Pernicious anemia | Primary biliary cirrhosis |
| Autoimmune cytopenia | Diabete mellitus type I |
| Graves' Disease | Rheumatoid arthritis |
| Systemic lupus erythematosus | Multiple sclerosis |
| Myasthenia gravis | Celiac disease |
| Bullous pemphigoid | |
| Pemphigus |
Related Talk
Kate Webb, Centre for Adolescent Rheumatology – Autoimmune diseases in children



