





- Patient presentation
- History
- Differential Diagnosis
- Examination
- Investigations
- Special investigations
- Discussion
- Treatment
- Case Final outcome
- Case Evaluation - Questions & answers
- MCQs
Patient presentation
A 9 year old girl presents with a one month history of generalised body swelling associated with shortness of breath and backache.
Acknowledgement
This case study was kindly provided by Dr Bhadrish J Mistry, Paediatrician, Department of Paediatrics, Chris Hani Baragwanath Hospital and the University of Witwatersrand.
Partnership
We have partnered with The International Union of Basic and Clinical Pharmacology (IUPHAR) to bring you in-depth information about drugs and pharmacology with links to the Guide to ImmunoPharmacology.
History
Two and a half weeks previously the patient had been admitted to her local hospital for a similar episode.
No other illnesses or admissions were noted.
Differential Diagnosis
- Nephrotic Syndrome
- Liver disease
- Cardiac Failure
- Protein Losing Enteropathy
- Malnutrition
Examination
On Admission- 20 February
She was found to have multiple naevi on her trunk, neck and face. Mostly small, dark brown, polygonal, irregularly shaped macules, 2-5 mm in diameter except for one large naevus on the right temple which was 3 cm in diameter.
Clinically she was in congestive cardiac failure which was complicated by:
- 2 large left ventricular thrombi (seen on echo, see investigations)
- Renal and liver dysfunction secondary to poor perfusion and congestion
Investigations
| Examination | Value | Normal Limits | |||||
|---|---|---|---|---|---|---|---|
| On Admission | 41703 | 41723 | 41752 | ||||
| 41690 | |||||||
| WBC | 12.7 | (4-12 x109/L) | |||||
| HB | 14.6 | (12.1-15.2 g/L) | |||||
| HCT | 0.43 | (.31-.42) | |||||
| Platelets | 138 | (140-450 x109/L) | |||||
| MCV | 94 | (80-100 fl) | |||||
| CRP | 8.1999999999999993 | ( | |||||
| NA | 126 | 129 | 136 | 137 | (135-147 mmol/L) | ||
| K | 5.9 | 6 | 4.3 | 4.2 | (3.5-5.1 mmol/L) | ||
| CL | 89 | 92 | 99 | 100 | (95-107 mmol/L) | ||
| CO2 | 10 | 30 | 25 | 26 | (22-33 mmol/L) | ||
| UREA | 20.9 | 11.7 | 8.8000000000000007 | 6.6 | (2.5-6.7 mmol/L) | ||
| CREAT | 74 | 51 | 37 | 42 | (70-150 µmol/L) | ||
| TBIL | 91 | 16 | (3-17 µmol/L) | ||||
| INDIR BIL | 58 | 6 | (0-6 µmol/L) | ||||
| TPROT | 48 | 60 | (60-80g/L) | ||||
| ALB | 30 | 35 | (35-50g/L) | ||||
| ALP | 208 | 78 | (30-150 U/L) | ||||
| GGT | 269 | 551 | (11-51 U/L) | ||||
| ALT | 468 | 17 | (5-35 U/L) | ||||
| AST | 1287 | 28 | (5-35 U/L) | ||||
| CK | 9663 | 25-195 iu/L | |||||
| CKMB | 616 | ||||||
| Troponin T | 0.16 | ||||||
| ANA | negative | ||||||
| C3 | Treatment | Treatment | 1.24 | 1.18 | (0.5-1.53) | ||
| C4 | 0.191 | 0.27800000000000002 | 0.251 | (0.2-1) | |||
| with | with IV | ||||||
| Coxsackie | |||||||
| Immunoglobulin | Methylprednisone | ||||||
| Viral Titres- | |||||||
| IgG | |||||||
| B1 | |||||||
| B2 | 40 | 160 | |||||
| B3 | 10 | ||||||
| B4 | >640 | 320 | |||||
| B5 | 320 | 80 | |||||
| B6 |
Special investigations
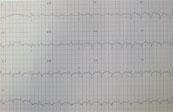
Echocardiography
Showed dilated cardiomyopathy with a shortening fraction of 9% (SF 28-44%) with pulmonary hypertension and 2 large left ventricular thrombi.
ECG
- R axis deviation
- P pulmonale
- Small wide QRS complexes
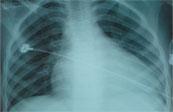
Chest X-Ray
Cardiomegaly
Skin Biopsy
Skin biopsy of a naevus was reported as an intradermal congenital melanocytic naevus
Discussion
Based on the echo and blood results the cause is now considered to be:
- viral myocarditis, or
- LEOPARD syndrome, a complex dysmorphogenetic disorder of variable penetrance and expressivity. The acronym LEOPARD is used to describe the main features of the disorder:
Lentigines (multiple)
- Electrocardiographic conduction abnormalities
- Ocular hypertelorism
Pulmonary stenosis
- Abnormalities of genitalia
- Retardation of growth
- Deafness
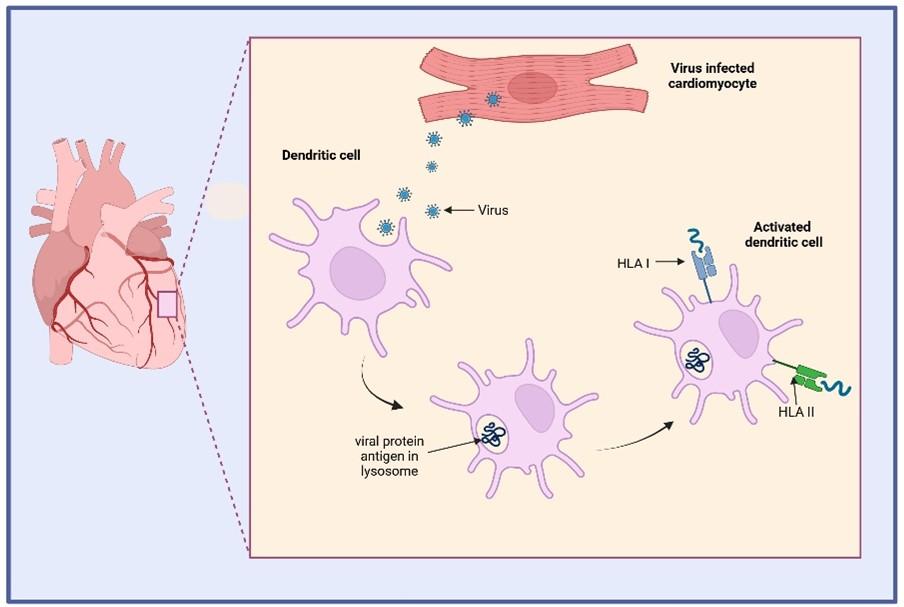
Figure 1 Autoimmune myocarditis is thought to be precipitated by an initial infection with a pathogen, such as a virus, that expresses surface B-cell epitopes highly similar to host epitopes found on the cell surface of cardiomyocytes (molecular mimicry). Viruses with tropism for myocardiocytes such as Adenovirus and Coxsackie B viruses are frequently associated with viral myocarditis. In a model for viral myocarditis, viral infection in the heart is first detected by the innate immune response involving phagocytes (macrophages and dendritic cells) that engulf virus and viral antigens released from infected cells. Viral peptide antigens are processed by dendritic cells and bound to HLA class I and II receptors for presentation to CD4+ and CD8+ T cells in regional lymph nodes. Created in https://BioRender.com (CA Petersen 2025)
The discussion for this case study will focus on the immunological process of viral myocarditis.
Myocarditis is an inflammatory disorder of the myocardium with necrosis of the cardiomyocytes and associated inflammatory infiltrate. It is usually caused by viruses which have tropism for myocardiocytes with the two most come viruses being adenovirus and coxsackie B viruses, which infects cariomyocytes via the coxsackievirus and adenovirus receptor (CAR)(Won et al., 2024). These viruses are therefore frequently associated with viral myocarditis. (see investigations)
Myocarditis generally results in a decrease in myocardial function, with accompanying cardiac enlargement and an increase in the end-diastolic volume due to increased preload. Normally, the heart compensates for dilation with an increase in contractility (Starling law), but because of inflammation and muscle damage, the heart is unable to respond to the increase in volume.
Dilated cardiomyopathy (DCM), is idiopathic in most cases and is a primary myocardial disorder characterised by cardiac enlargement and impaired systolic function of one or both ventricles. Despite conventional therapies for DCM the prognosis remains poor. There is increasing evidence that suggests that myocarditis and dilated cardiomyopathy are closely linked. Autoimmunity is one of the main mechanisms in the pathogenesis that links myocarditis to DCM.
Viral myocarditis, is first detected by the innate immune response involving phagocytes macrophages and dendritic cells) that engulf virus and viral antigens released from infected cells. Viral peptide antigens are processed by dendritic cells and are bound to HLA class I and II receptors for presentation to CD4+ and CD8+ T cells in regional lymph nodes.
Dendritic cells present peptide antigens bound to surface HLA class II receptors to naive CD4+ helper T lymphocytes in the T cell zone of lymph nodes. CD4+ T cells expressing T cell receptors recognise antigen and become activated and proliferate into effector and memory CD4+ T cell populations.
Dendritic cells also present peptide antigens bound to surface HLA class I receptors to naive CD8+ cytotoxic T lymphocytes in the lymph nodes. These CD8+ T cells then receive activation signals from CD4+ helper T lymphocytes and become primed for cell-killing. Once activated they differentiate into effector or memory CD8+ T cells with effector cells able to kill host cells bearing viral peptide antigens.
In viral myocarditis the cardiomyocytes which are infected with virus express surface HLA class I receptors with bound viral peptide antigens. Effector CD8+ cytotoxic T lymphocytes that express T cell receptors able to recognise viral peptide antigens actively kill infected cells.
Virus-specific antibodies are synthesised by plasma B cells following priming and activation of virus-specific B lymphocytes.
The CD4+ helper T lymphocytes which recognise antigen bound to HLA class II receptors on B cells become activated and in turn activate primed B cells to differentiate into effector plasma B cells that secrete antibodies and memory B cells. Naive B cells initially synthesise IgM while re-stimulated memory B cells are able to isotype-switch depending on the type of activation signals obtained from CD4+ helper T cells.
Virus-specific antibodies function to neutralise and opsonise free virus particles. Virus and viral antigens opsonised with antibodies also form immune complexes which can be detected and destroyed by phagocytes expressing Fc receptors. Furthermore the classical complement cascade can augment this response by opsonising virus and viral antigens with C3b complement.
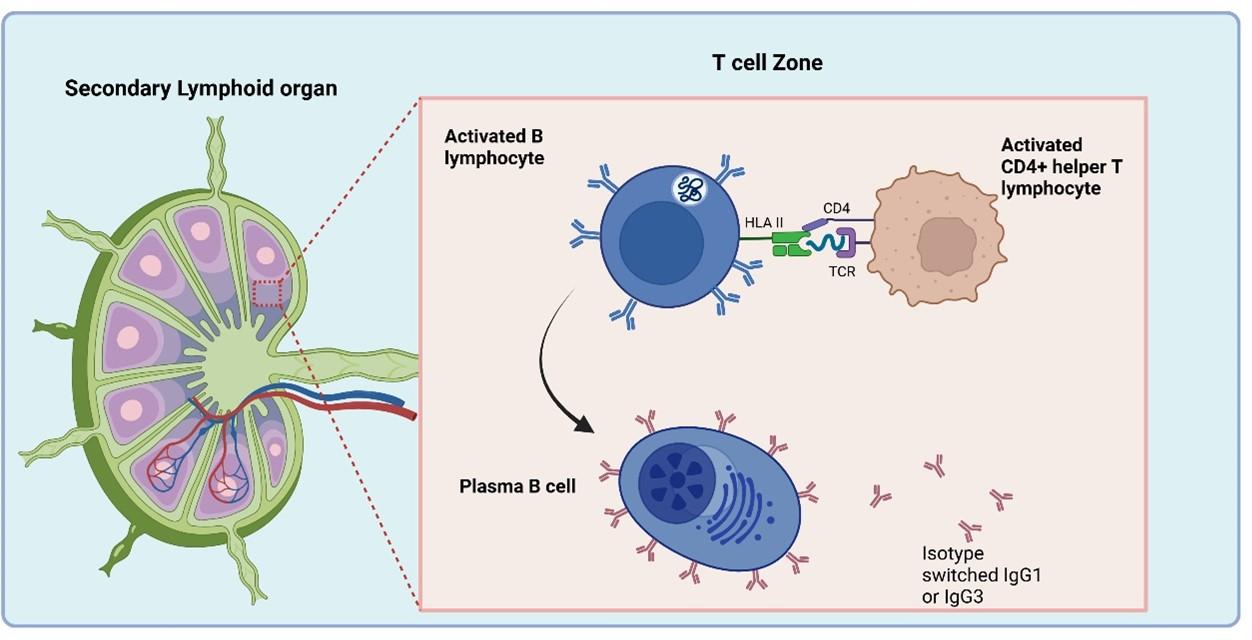
Figure 2 Dendritic cells present peptide antigens bound to surface HLA class II receptors to naive CD4+ helper T lymphocytes in the T cell zone of secondary lymphoid tissue (lymph nodes). CD4+ T cells that express T cell receptors that recognise antigen become activated and proliferate into effector and memory CD4+ T cell populations. Effector CD4+ T cells migrate to inflamed tissue where they further stimulate the immune response. Created in https://BioRender.com (CA Petersen 2025)
Autoimmune disease as a consequence of viral myocarditis is thought to be mediated by virus-specific antibodies previously synthesised that cross-react with a surface protein expressed on cardiomyocytes (molecular mimicry). Antibodies of the IgM and IgG class can activate the classical complement cascade which leads to lysis of healthy cells. Triggering the classical complement pathway (link to complement classical pathway drawings) also results in consumption of C3 and C4 (see investigations). Natural killer cells and phagocytes bearing Fc receptors may also play a role in the destruction of antibody-bound host cells.
Destruction of healthy cardiomyocytes results in the release of large quantities of intracellular antigens that are engulfed by macrophages. Macrophages present self-peptides to CD4+ T helper cells which results in a breakdown in tolerance of auto-antigens leading to the generation of autoreactive CD4+ helper T cells. These CD4+ helper T cells that can provide activation signals to autoreactive B cells. In this way secondary antibodies directed against intracellular host antigens derived from the heart can develop. These antibodies include anti-nucleic acid, anti-myosin and anti-actin which are detectable in severe autoimmune myocarditis.
Treatment
- IV Immunoglobulin was given after the first C3 and C4 result was measured. Subsequent C3 and C4 results were normal.
- Methylprednisone was given after one month because there was deterioration in the cardiac function of this patient and worsening ascites and respiratory distress. Shortly after treatment a rise in coxsackie B2 titre was seen, which was thought to be due to immunosuppression.
Final outcome
- Patient developed a possible gastritis or peptic ulcer disease and was commenced on omeprazole.
- She later developed hypovolaemic or cardiogenic shock and was transfused with packed red cells and treated with inotropes.
- This was followed by a gradual resolution of renal & liver dysfunction.
- The two left ventricular thrombi were resolved with warfarin.
- Cardiac function improved from SF 9% to SF 19%.
Evaluation – Questions & answers
What is the diagnosis?
What is the cause of viral myocarditis?
What is autoimmune myocarditis?
What is molecular mimicry?
How is viral infection in the heart first detected?
What is the process for antibody secretion?
Multiple Choice Questions
Earn 1 HPCSA or 0.25 SACNASP CPD Points – Online Quiz



