





- Patient presentation
- History
- Differential Diagnosis
- Examination
- Investigations
- Discussion
- Treatment
- Final outcome
- Evaluation - Questions & answers
- MCQ
- References
Patient presentation
An 8 year old boy is referred to a regional hospital for a surgical consultation for a lobectomy following a diagnosis of bronchiectasis due to a chronic history of recurrent respiratory infections.
Acknowledgement
This case study was kindly provided by Dr Monika Esser MMed Paed, Head of Division of Immunology, N.H.L.S Coastal Branch, Tygerberg Hospital.
History
- Bacterial meningitis at age 2 years complicated by deafness – for which he currently attends a school for hearing impaired children.
- Pulmonary TB fully treated, exact age at time of treatment unknown.
- Currently presents with bronchiectasis due to recurrent respiratory infections.
- No significant family history.
Differential Diagnosis
- HIV
- X- linked Agammaglobulinaemia (Bruton’s Disease)
- Agammaglobulinaemia
- Hyper IgM Syndrome
- Common variable immunodeficiency
- Severe Combined Immunodeficiency
Examination
On general examination:
- Does not appear acutely ill
- Apyrexial
- Weight on 75th centile, height 90th centile
- Clubbing of fingernails
Respiratory System:
- Persistent productive cough
- Harrison’s sulcus
- Bilateral lower lobe crepitations
Other systems:
- Nil of note
Investigations
| FBC: | |
|---|---|
| WBC | 11.11 x 10’9/L – 81.3% Lymphocytes |
| Hb | 12.5 g/L |
| Platelets | 397 x 10’9/L |
| (no other counts available) |
| Sputum: | |
|---|---|
| H. Influenza and | Isolated on culture |
| S. Pneumoniae | |
| TB Smear | Negative |
| TB Culture | Negative |
| CXR | Suggestive of bronchiectasis with superimposed infection |
| Serum Immunoglobulins: | |||
|---|---|---|---|
| Se | IgG | 0.393 g/L | (6-20) |
| IgA | (0.8-3) | ||
| IgM | 1.09 g/L | (0.4-1.8) | |
| IgE | 4.3 IU/ml | (>60 IU/ml) |
| Lymphocyte Subset Analysis: | ||
|---|---|---|
| Percentage | Absolute | |
| CD19 1% (L) | 90 (L) | |
| CD3 97% (H) | 8759 (H) | |
| CD4 22% (L) | 1987 (H) | |
| CD8 68% (H) | 6140 (H) | |
| NK 8% (L) | 722 (H) | |
| Ratio 0.32:1 | ||
| Lymphocyte subsets showed low but not entirely absent CD19 (B cells) and elevated C3 and NK cells. |
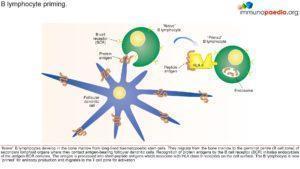 Discussion
Discussion
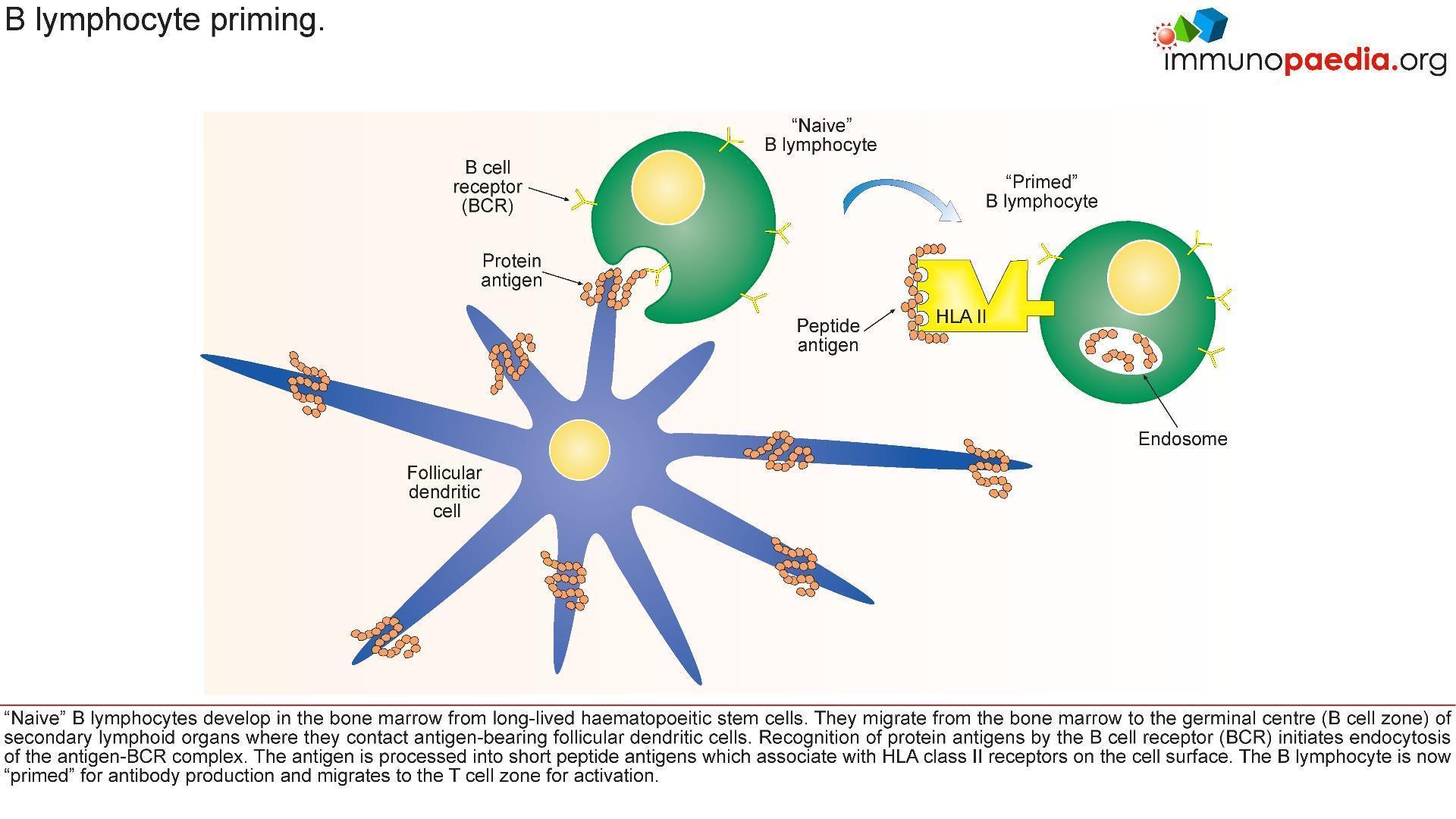
- Upon investigation it was noted that the IgA, IgE and IgG were considerably lower than that of IgM. This was an indication of defective class switching recombination process making Hyper IgM syndrome the evident diagnosis.
- Hyper IgM syndrome is a disease that causes a defective interaction of CD40L(CD154)- CD40 between CD4+ T cells and antigen presenting cells.
- Hyper IgM syndrome patients generally indicate decreased number of isotope memory B cells along with normal numbers of total B cells.
- The majority of patients affected usually presents with a broad spectrum of clinical manifestations which usually develops during infancy and second year of life.
This makes them susceptible to recurrent bacterial (meningitis, pneumonia, and otitis amount other) and opportunistic infections. Patients diagnosed with Hyper IgM syndrome are prone to pulmonary complication, gastrointestinal manifestations, autoimmune disorder, hematologic abnormalities, lymphoproliferation disorders and malignancies (Vaillant & Qurie, 2023; Yazdani et al., 2019)
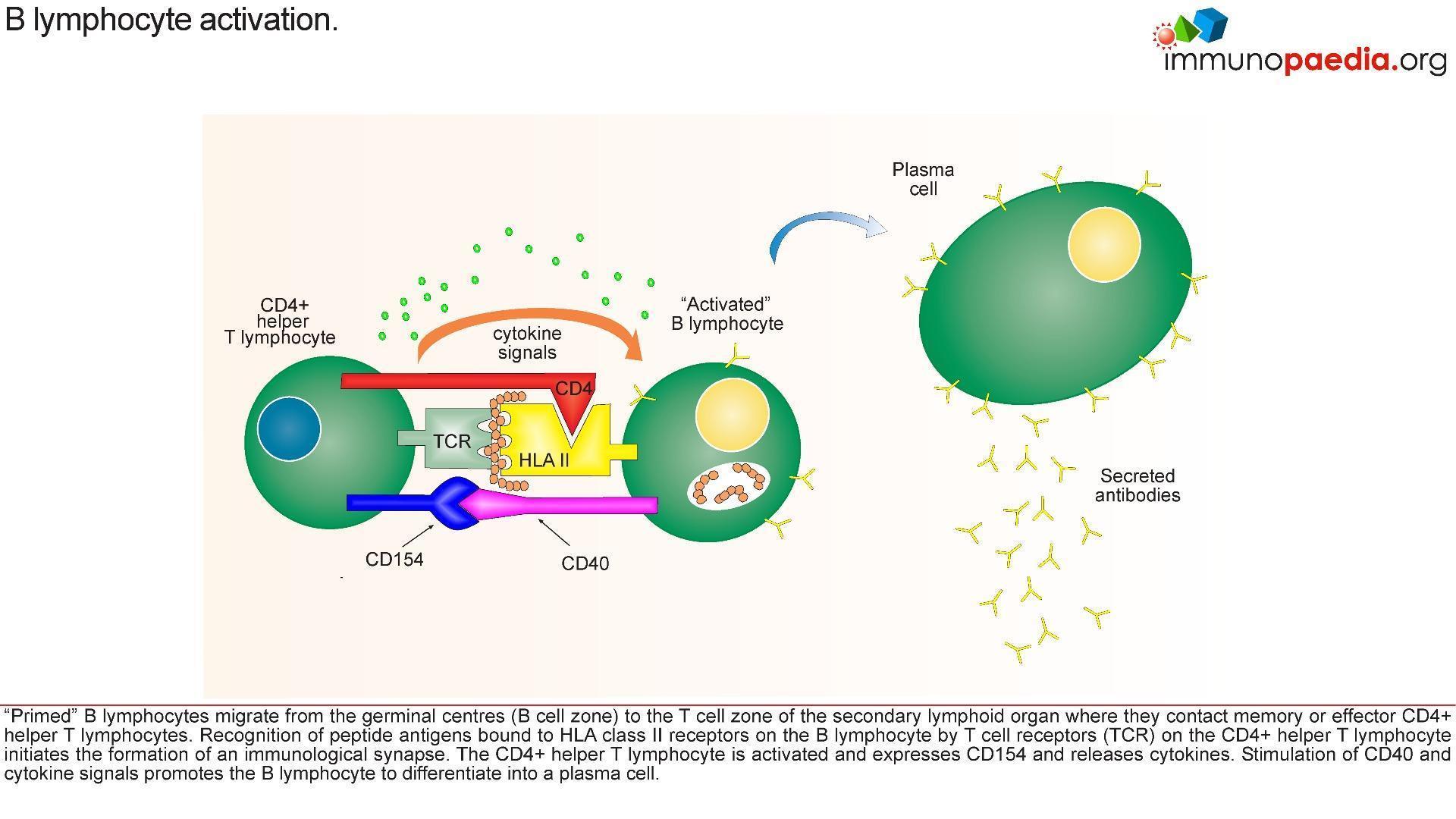
The Role of CD40 and its ligand (CD154)
- The CD40L gene is located on the X chromosomes which encodes CD40L, is expressed on activated T cells and is a member of the tumour necrosis factor (TNF) family.
- The mutation in this gene usually affects the extra cellular domain, resulting in a defective geometrical fold of the CD40L or preventing the CD40L/CD40 binding.
- CD40, is expressed by a variety of cells including B cells, macrophages, dendritic cells and other nonimmune cell types.
- CD40 activation occurs via binding to its ligand CD154 (CD40L), which is expressed on activated T cells
- However, a mutation in the CD40 or CD154 leads to improper binding and a defective T cell co-stimulation, causing a combined T (cellular) and B (humoral) cell immunodeficiency. which is expressed on activated T cells.
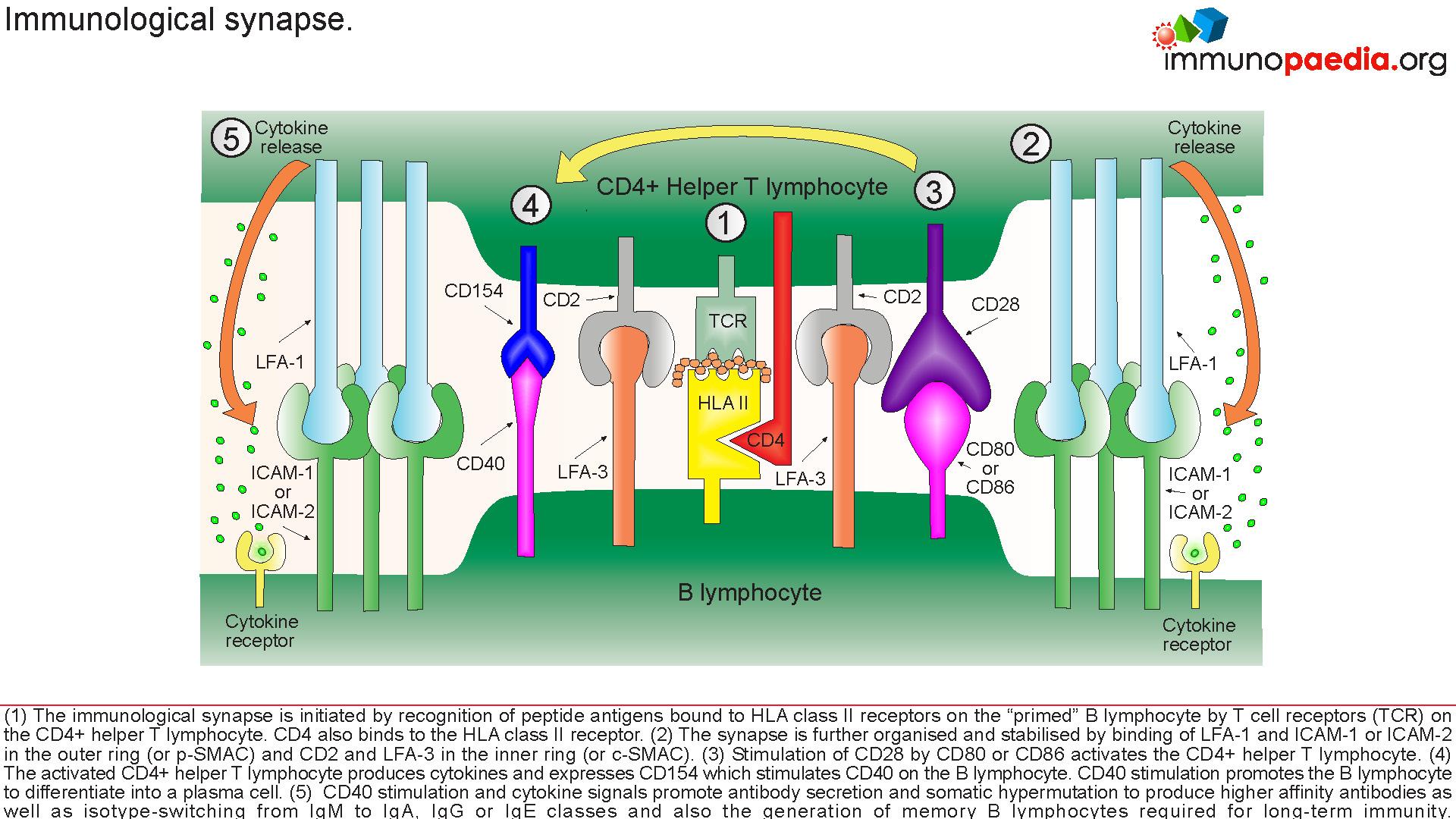
CD40 activation is critical for:
- B cell proliferation
- Ig- Isotype switching
- Germinal centre formation
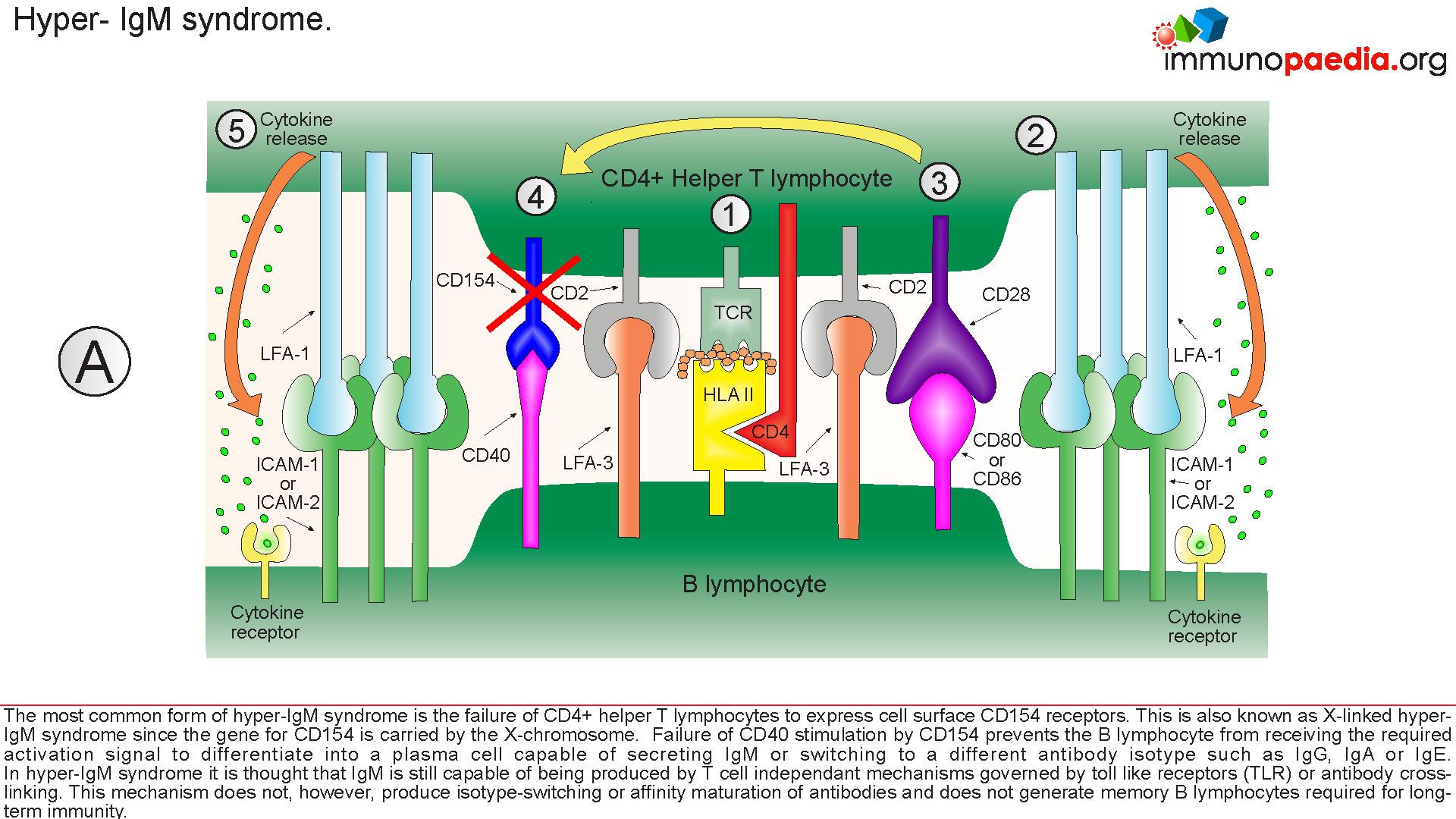
Mutations in either CD40 or CD154 gene cause Hyper IgM Syndrome , HIGM3 and HIGM1 respectively , which is characterised by:
- Normal or elevated SeIgM
- Markedly decreased IgG, IgA, IgE

Due to lack of CD154, T cell interaction with protein antigen processing cells such as CD19 is impaired preventing ligand interaction of B cell CD40 with CD154 on T cells with subsequent defective response to T cell dependent antigens i.e. the immunological synapse is not formed.
This synapse gives essential costimulatory signals to B cells, necessary for the B cells to switch from low affinity IgM production primary response to B cell producing high affinity antibodies of different classes such as IgG and IgA. 65-75% of the cases observed, has shown a correlation of Hypo IgM syndrome to an X linked CD40 recessive autosomal mutation, that mostly affect male. However, there are other genetic mutations that also leads to Hypo IgM syndrome:
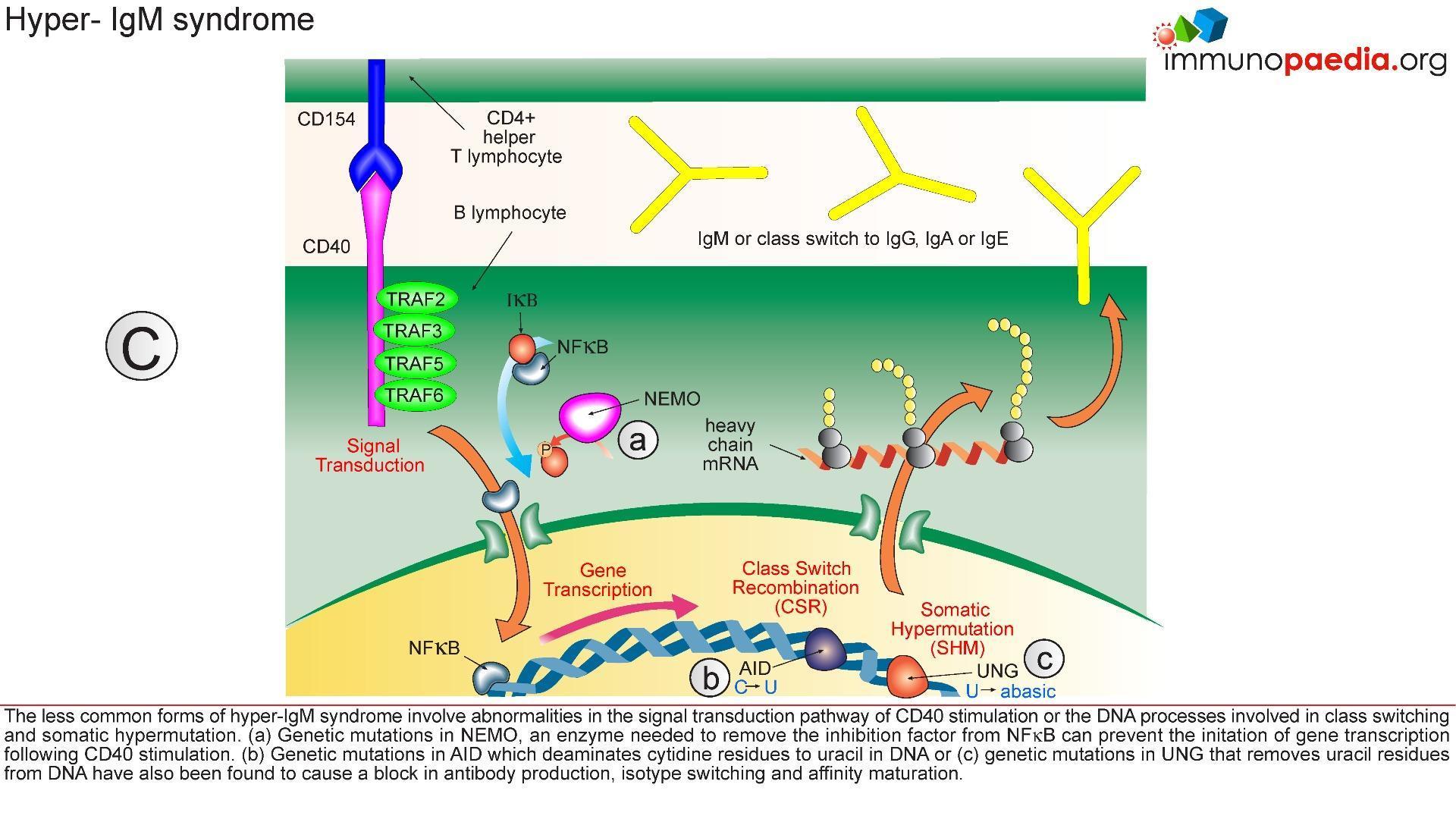
- AICDA and UNG mutations
- PI3K and PI3KCD mutations
- NEMO and IKBA mutations
- DNA repair gene mutations
Download images for this case
Treatment
- Physiotherapy daily. After 4 weeks of intensive physio a CT chest confirmed bilateral basal bronchiectasis.
- Broad spectrum antibiotics were initiated.
- IVIG at 0,4g/kg – 3 weekly infusions. (Antibody studies to common vaccine antigens were not done prior to IVIG therapy and are therefore not available).
Download images for this case
Final outcome
Long-term Plan:
- IVIG infusions 3 weekly to maintain normal serum levels of IgG and prompt treatment with antibiotics for bacterial infections.
- Prevention or early recognition of protozoal infections of liver and GIT.
- Cotrimoxazole prophylaxis for PCP infections.
- Bone marrow transplant would have been the curative treatment of choice, prior to established bronchiectasis.
Download images for this case
Evaluation – Questions & answers
What is the diagnosis?
Give possible reasons for bronchiectasis developing in childhood.
What else on history is essential for evaluating this patient with regards to immunity?
Which other clinical findings would you like information on?
If this child has a primary or secondary Immunodeficiency - which component of the immune response do you expect to be impaired?
Comment on the immunoglobulin levels and lymphocyte phenotypes.
How do you explain a normal Se IgM with the other Immunoglobulin isotypes low?
Could this patient have X-linked Agammaglobulinaemia (XLA) (Bruton’s)?
What is the possible cause of the T cell subset abnormal distribution?
How would you expect a patient with very low B cells to respond to vaccination?
Who in the boy’s family would possibly be the best match for HLA typing for a bone marrow transplant?
Download images for this case
Multiple Choice Questions
Earn 1 HPCSA or 0.25 SACNASP CPD Points – Online Quiz
Download images for this case
References
- Vaillant, A. A. J., & Qurie, A. (2023). Immunodeficiency. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK500027/
- Yazdani, R., Fekrvand, S., Shahkarami, S., Azizi, G., Moazzami, B., Abolhassani, H., & Aghamohammadi, A. (2019). The hyper IgM syndromes: Epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clinical Immunology, 198, 19–30. https://doi.org/10.1016/J.CLIM.2018.11.007



