





Pancreatic cancer remains one of the most aggressive and treatment-resistant malignancies, with few effective therapeutic options and a poor prognosis for most patients. A new study has uncovered an unexpected vulnerability, one rooted in how cancer cells exploit their own damaged mitochondria to survive (Figure 1).
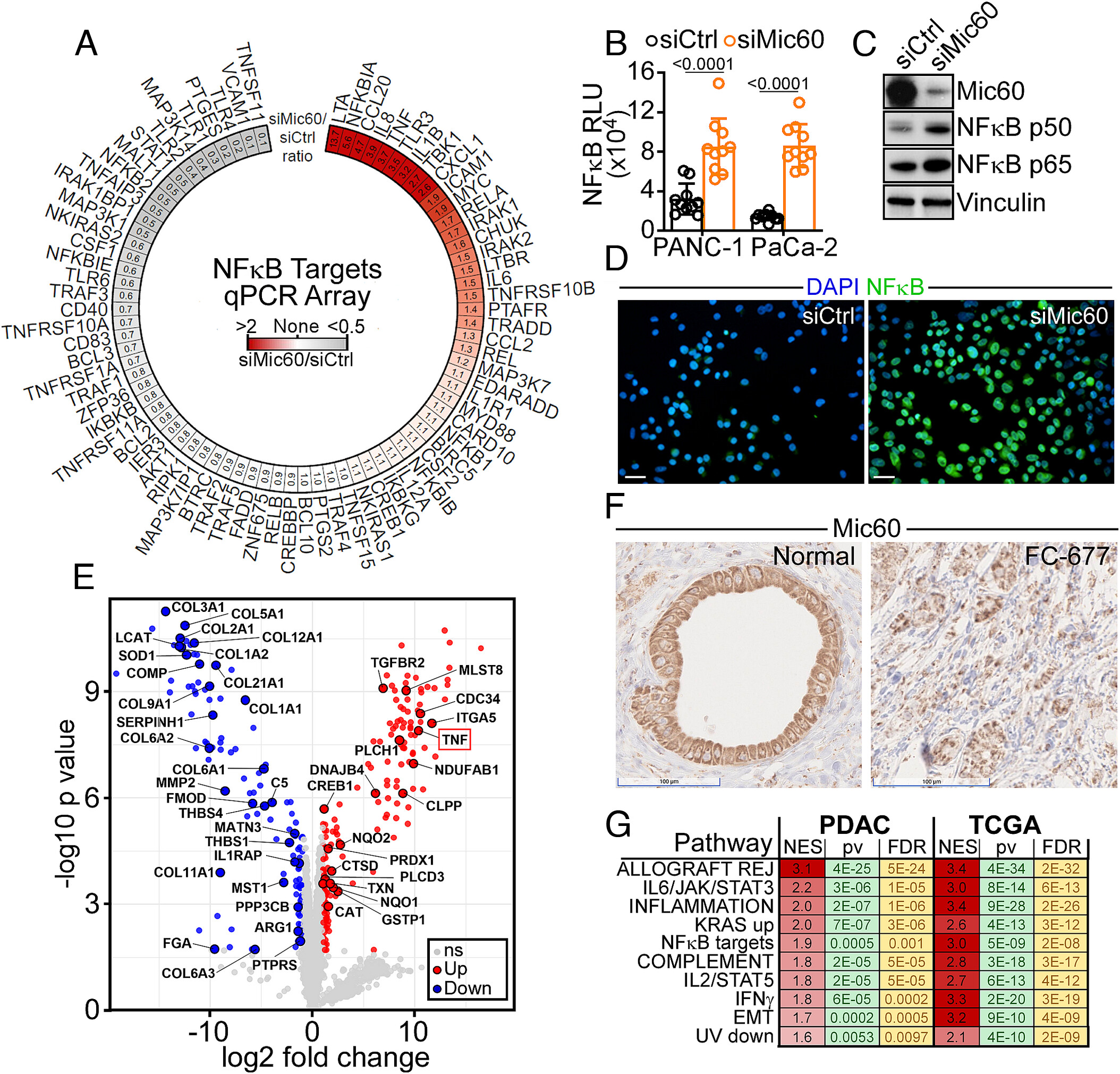
Figure 1: NFκB inflammation in PDAC. (A) PANC-1 cells expressing control nontargeting siRNA (siCtrl) or Mic60-directed siRNA (siMic60) were analyzed in a 78-gene NFκB profiler array by RT-qPCR. Data are expressed as siMic60/siCtrl ratio in a radial heatmap with the ratio magnitude highlighted by color. Representative experiment. (B) PANC-1 or MIA PaCa-2 cells expressing siCtrl or siMic60 were analyzed for NFκB promoter reporter activity. RLU, relative luciferase units. Mean ± SD. Numbers are P values by two-tailed unpaired t test. (C and D) PANC-1 cells as in (B) were analyzed by Western blotting (C) or confocal fluorescence microscopy for nuclear localization of p65 NFκB (D). Representative images. (Scale bar, 20 µm.) (E) Aliquots of conditioned medium harvested from PANC-1 cells as in (B) were analyzed by mass spectrometry and 298 differentially secreted proteins were identified. Data are presented as a volcano plot showing the log2 fold change of siMic60 compared to control. TNFα released in the Mic60-knockdown secretome is indicated. ns, not significant. (F) Pancreas tissue samples from two patients with histological diagnosis of PDAC were analyzed for Mic60 expression, by immunohistochemistry. Mic60 reactivity with an adjacent normal pancreatic gland (Normal) is shown. (Scale bars, 100 µm.) (G) Overlap of significantly enriched hallmark pathways between eight patients with histologic diagnosis of PDAC (PDAC) and PDAC patients in the TCGA (TCGA) dataset that were significantly positively associated (FDR < 5%) with an NFκB target gene signature. For each pathway, a normalized enrichment score (NES), P value (pv), and false discovery rate (FDR) are indicated.
The the work reveals a surprising mechanism: pancreatic tumours depend on self-generated inflammation triggered by dysfunctional mitochondria and blocking this pathway can kill the cancer cells.
Mitochondria are best known as the cell’s energy producers, but they also play key roles in stress signalling. In many pancreatic tumours, mitochondria are structurally defective due to reduced levels of a protein called Mic60. These damaged organelles, sometimes referred to as “ghost mitochondria”, lose membrane integrity.
This breakdown has a critical consequence:
they begin leaking double-stranded RNA (dsRNA) into the cytoplasm.
Normally, dsRNA is a red flag associated with viral infection. When cells detect it, they activate innate immune pathways designed to trigger inflammation and antiviral defence.
The leaked mitochondrial RNA is sensed by immune receptors, including Toll-like receptor 3 and its downstream signalling partner TRAF6.
This activates a powerful inflammatory cascade.
But instead of harming the tumour, this response is hijacked:
- Cancer cells use inflammation to support growth
- Survival pathways are amplified
- The tumour microenvironment becomes more permissive
In effect, the cancer creates a false viral alarm and then exploits the resulting inflammation to thrive.
Perhaps the most striking finding is that pancreatic cancer cells don’t just benefit from this inflammation, they become dependent on it.
When researchers blocked the TLR3/TRAF6 signalling pathway:
- Tumour cells lost their survival signals
- Cancer growth halted in mouse models
- Cancer cells died, while healthy cells remained largely unaffected
This suggests a classic case of non-oncogene addiction, where tumours rely on a stress-response pathway rather than a traditional mutation-driven driver.
Targeting the TLR3/TRAF6 axis could represent a completely new strategy for treating pancreatic cancer:
- It exploits a tumour-specific vulnerability
- It avoids directly targeting essential cellular processes in healthy tissue
- It may be applicable to other cancers with similar mitochondrial defects
Importantly, this is the first time this mitochondrial RNA–driven inflammatory pathway has been identified as a driver of cancer progression, rather than just a byproduct of cellular stress.
Inflammation has long been recognized as a double-edged sword in cancer—capable of both suppressing and promoting tumour growth. This study adds a new layer of nuance:
- Tumours can generate their own inflammatory signals internally
- Damaged organelles like mitochondria can act as signalling hubs, not just dysfunctional bystanders
- Blocking inflammation, in the right context, can directly impair tumour survival
For a disease as difficult to treat as pancreatic cancer, identifying a built-in weakness like this is significant. It shifts the strategy from trying to overpower the tumour to cutting off a process it has come to rely on for survival.
Journal article: Milcarek, A.T., et al. 2026. Mitochondrial double-stranded RNA fuels pancreatic cancer growth via RIG-I/TLR3 inflammation. PNAS.
Summary by Stefan Botha



