





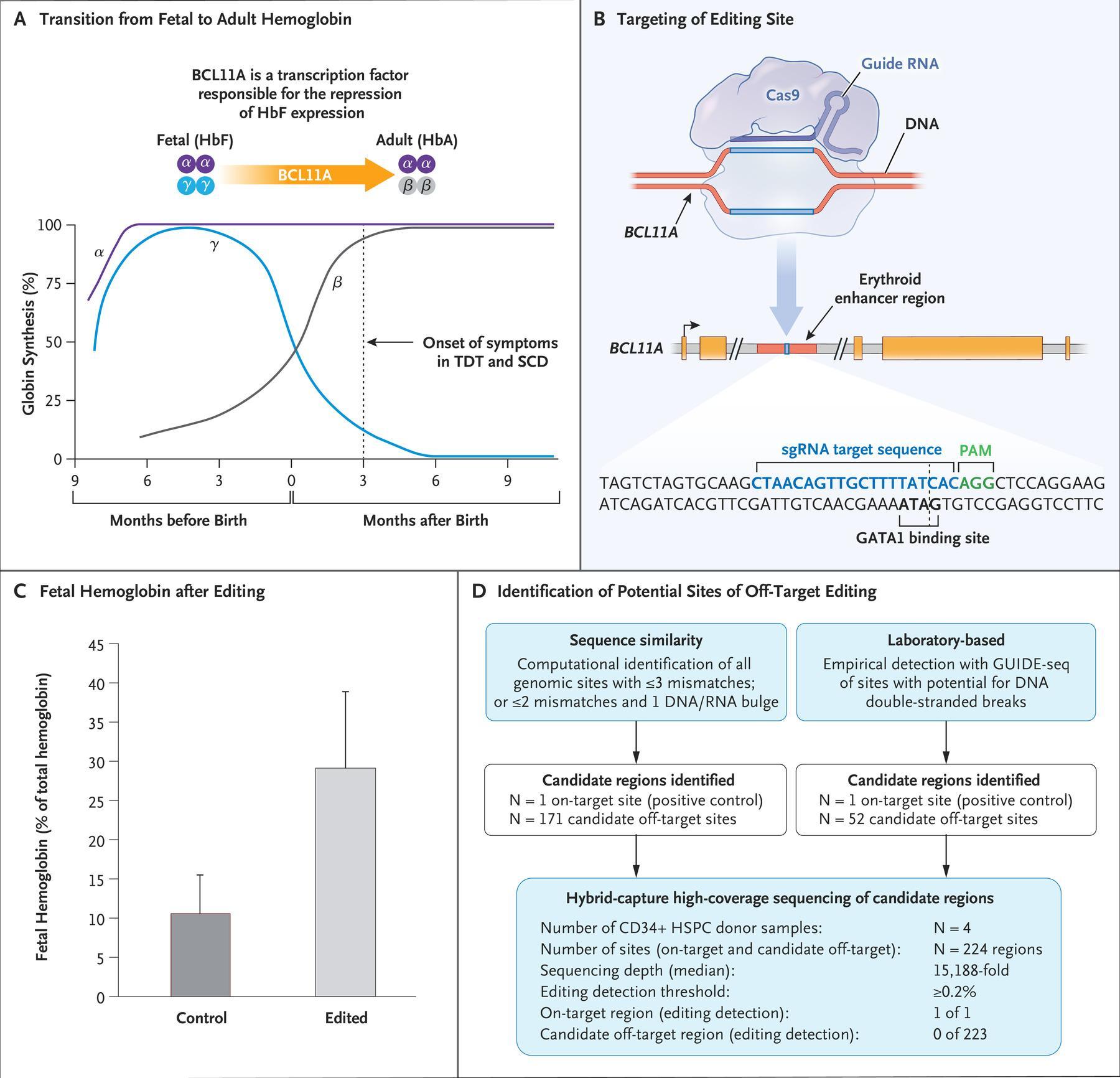
Panel A shows the transition from fetal hemoglobin (HbF) to adult hemoglobin (HbA) shortly after birth, along with the role of the transcription factor BCL11A in mediating the repression of γ-globin, a component of fetal hemoglobin. In patients who lack the ability to produce functional or sufficient β-globin, the onset of symptoms occurs coincident with the decrease in fetal hemoglobin levels approximately 3 months after birth. SCD denotes sickle cell disease, and TDT transfusion-dependent β-thalassemia. Panel B shows the target editing site of the single guide RNA (sgRNA) that directs CRISPR-Cas9 to the erythroid-specific enhancer region of BCL11A. (The sgRNA sequence is provided in the Supplementary Appendix.) The five BCL11A exons are depicted as gold boxes. GATA1 denotes the binding site of the GATA1 transcription factor, and PAM the protospacer adjacent motif (NGG, a specific DNA sequence required to immediately follow the Cas9 target DNA sequence). Panel C shows preclinical data regarding fetal hemoglobin as a percentage of total hemoglobin after editing and the differentiation of erythroid cells. Data are from samples obtained from 10 healthy donors. Error bars indicate the standard deviation. Panel D shows the results from off-target evaluation. GUIDE-seq denotes genomewide unbiased identification of double-strand breaks enabled by sequencing, and HSPC hematopoietic stem and progenitor cell. To nominate sites, GUIDE-seq was performed once independently on each of 3 CD34+ HSPC healthy donor samples. To confirm sites, hybrid capture was then performed on each of 4 CD34+ HSPC healthy donor samples. On-target allelic editing was confirmed in each experiment with a mean of 57%, and no detectable off-target editing was observed at any of the sites nominated by GUIDE-seq and by sequence homology. Panel A was adapted with permission from Canver and Orkin. (Source: Frangoul et al., 2020)
Sickle cell disease (SCD) is a genetic disease characterised by severe anaemia. Individuals with sickle cell have a single mutation in the haemoglobin β subunit gene (HBB) which causes deformation of red blood cells (RBCs), specifically RBCs in individuals with SCD have a sickle shape instead of a biconcave shape. These “sickled” RBCs have reduced capacity to carry oxygen compared to normal RBCs, are very “sticky” and tend to cause blockages in small blood vessels and capillaries leading to pain and progressive organ damage.
An estimated 300000 individuals are diagnosed with SCD annually. SCD is treated using pain management, blood transfusions (to increase normal RBC levels) and hydroxyurea (if available). Hydroxyurea increases fetal haemoglobin* levels resulting in higher oxygen-carrying capacity of RBCs in SCD patient (Platt et al., 1984 ;Rodgers et al., 1993), thus reducing pathology. Further, the only functional cure for SCD is via bone marrow transplants. However finding a suitable donor is very challenging (Eapen et al., 2019). Two studies published in December 2020 describe gene editing approaches to treat SCD, both studies aimed to increase the levels of fetal haemoglobin by silencing the BCL11A, a gene that represses expression of fetal haemoglobin in adult RBCs. Frangoul et al., (n=2) and Erisk et al., (n=6) utilised CRISPR-Cas9 gene editing and lentiviral vector-mediated RNA interference (RNAi), respectively, to silence BCL11A in autologous CD34+ hematopoietic stem and progenitor cells. Cells with the silenced BCL11A gene were transferred to patients with SCD and transfusion-dependent β-thalassemia# (Study by Frangoul et al., only). Both studies demonstrated the safety of gene editing and successfully increased fetal haemoglobin which led to a reduction in SCD morbidity. Unfortunately, this immunotherapy is still costly and challenging to conduct. Further, long term safety and durability of the “functional cure” are yet to be determined as the maximum median follow-up of any individual in either study was less than 2 years. Despite the small sample size, results from these studies are very promising and suggest potential utility in gene editing of own cells as a cure for a genetic disease.
*Production of fetal hemoglobin is developmentally regulated so that the level of γ-globin that is produced in utero decreases postnatally as the production of β-globin and adult hemoglobin (consisting of two alpha and two beta chains) increases. Fetal hemoglobin has higher binding affinity to oxygen than adult hemoglobin
# Transfusion-dependent β-thalassemia a disease caused by mutations in the haemoglobin β subunit gene which results in in reduced or β -globin synthesis and an imbalance between the α-like and β-like globin (e.g., β, γ, and δ) chains of hemoglobin, which causes ineffective erythropoiesis.
Journal Articles:
- Erisk et al., 2020. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. NEJM
- Frangoul et al., 2020. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. NEJM
Summary by Cheleka AM Mpande
Listen to the following podcast to get a general information on Sickle Disease.



