





A new study provides one of the most detailed explanations yet for why therapies targeting IL-23 can produce long-lasting remission in psoriasis, even after treatment has stopped (Figure 1). By combining single-cell RNA sequencing with spatial transcriptomics, researchers mapped the cellular and molecular changes occurring in psoriatic skin before and after IL-23 blockade, revealing how these therapies dismantle the inflammatory networks that sustain disease.
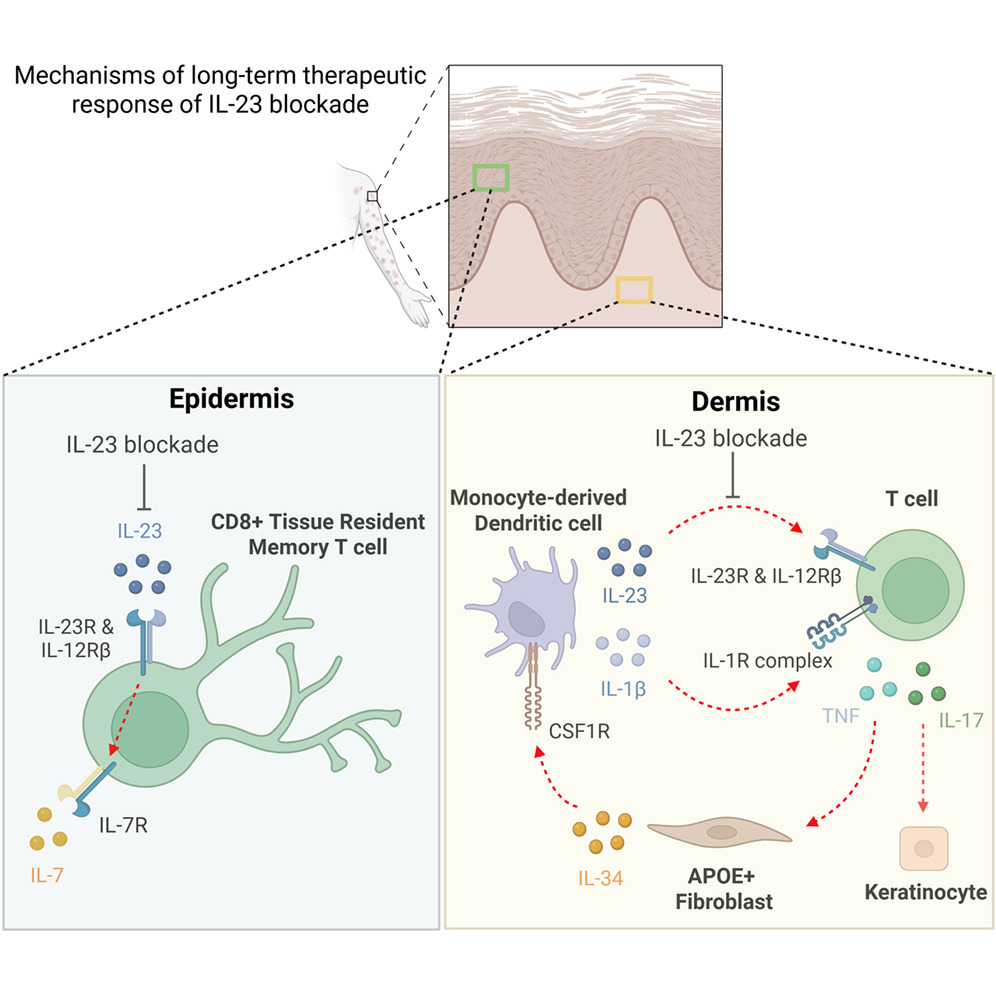
Figure 1: Graphical abstract.
Psoriasis is a chronic immune-mediated skin disorder driven largely by the IL-23/IL-17 inflammatory axis. IL-23 promotes the expansion and maintenance of IL-17-producing T cells, which drive keratinocyte activation, inflammation, and the formation of characteristic psoriatic plaques. While biologics targeting IL-23 have shown remarkable clinical success, many patients continue to experience disease control long after treatment cessation, suggesting that these therapies induce more profound immune remodelling than simple cytokine suppression.
To understand these long-term effects, researchers analysed matched skin biopsies from patients before and after IL-23 blockade and followed them for 36 weeks after treatment withdrawal. Their findings reveal that IL-23 inhibition fundamentally reshapes the immune ecosystem of psoriatic skin by targeting several interconnected cellular circuits that maintain chronic inflammation.
One of the most striking effects was observed in CD8⁺ tissue-resident memory T cells (TRM cells), which are increasingly recognized as key drivers of psoriasis recurrence. These cells persist within previously affected skin and can rapidly reignite inflammation after treatment ends. The study found that IL-23 blockade substantially reduced IL-17-producing resident memory CD8⁺ T cells and disrupted their long-term survival program.
Mechanistically, this effect was linked to suppression of a c-MAF–IL-7 survival axis. The transcription factor c-MAF and the cytokine IL-7 are critical for maintaining memory T cell persistence within tissues. By attenuating this pathway, IL-23 inhibition appears to weaken the ability of pathogenic resident memory T cells to survive long-term within the skin, potentially explaining the durable clinical responses observed after treatment discontinuation.
The study also identified an unexpected role for stromal cells in sustaining inflammation. A subset of fibroblasts characterized by expression of APOE and IL34 was significantly reduced following IL-23 blockade. These fibroblasts were found to produce IL-34, a cytokine that promotes the differentiation of infiltrating monocytes into inflammatory dendritic cells. As fibroblast-derived IL-34 levels declined, pro-inflammatory dendritic cells also decreased, interrupting another important source of inflammatory cytokines within psoriatic lesions.
This finding highlights an emerging concept in immunology: stromal cells are not passive structural elements but active regulators of immune responses. In psoriasis, fibroblasts appear to help sustain inflammatory circuits by supporting dendritic cell generation and function. IL-23 blockade therefore impacts not only immune cells directly but also the tissue microenvironment that supports chronic inflammation.
The researchers further discovered changes within the skin vasculature. Specialized post-capillary venules expressing the chemokines CCL19 and CCL21 normally help recruit T cells and other immune cells into inflamed tissue. Following IL-23 inhibition, expression of these chemokines was markedly reduced, resulting in decreased immune cell trafficking into the skin. This vascular remodelling likely contributes to the sustained reduction in inflammatory cell infiltration observed after treatment.
Taken together, the findings suggest that IL-23 blockade achieves durable disease control by simultaneously targeting multiple components of the psoriatic inflammatory network. Rather than simply reducing IL-17 production, the therapy weakens pathogenic resident memory T cells, suppresses inflammatory dendritic cell generation, alters fibroblast behaviour, and reduces immune cell recruitment through the vasculature. These coordinated changes effectively dismantle the self-sustaining inflammatory loops that drive chronic disease.
Beyond psoriasis, the study has broader implications for understanding tissue-resident immune memory in chronic inflammatory disorders. Resident memory T cells have been implicated in diseases ranging from inflammatory bowel disease to autoimmune arthritis, and strategies that selectively disrupt their survival may offer long-lasting therapeutic benefits without widespread immune suppression.
Journal article: Jiang, R., et al. 2026. A longitudinal atlas of human psoriatic skin reveals the mechanisms of anti-IL-23 therapy in disrupting the type 17 inflammatory circuit. Immunity.
Summary by Stefan Botha



