





Mycobacteria such as Mycobacterium tuberculosis can survive and multiply inside immune cells called macrophages, but how they achieve this has remained unclear. New research reveals that these bacteria hijack a host immune receptor to create a more permissive intracellular environment (Figure 1).
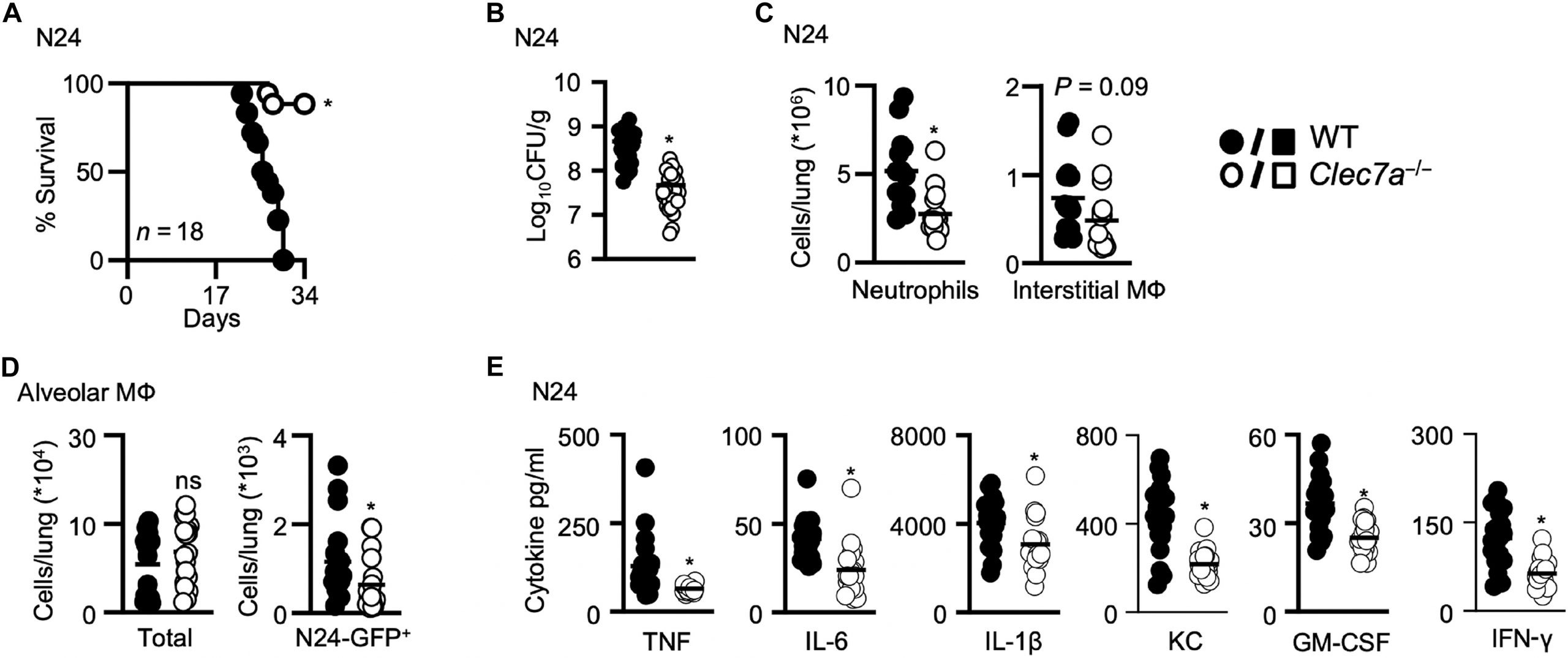
Figure 1: Dectin-1–deficient mice are resistant to mycobacterial infection. (A) Survival of WT (n = 18) and Clec7a−/− (n = 18) 129Sv mice after intranasal infection with the clinical strain MTB N24. Pooled data from two experiments (day 1 loading control: 400, 687 CFU). Pulmonary bacterial burdens (CFU, bar indicate average; n = 18) (B) and cellular composition (n = 18) (C) in the lungs of 129Sv WT and Clec7a−/− mice 21 days after infection with MTB N24. Neutrophils were defined as CD11b+Ly6G+ and interstitial macrophages (MФ) as CD11b+CD11c−SiglecF−F4/80+. (D) Total (left) and number of GFP+ alveolar macrophages (right) in the lungs of mice 14 days after intranasal infection with 400 to 600 CFU of MTB N24-GFP (n = 18). (E) Concentrations of inflammatory cytokines in lung homogenates at day 21 after infection with MTB N24 in WT and Clec7a−/− mice, as determined by Luminex cytokine profiling (n = 8). All data shown were pooled from at least two independent experiments, with bar charts showing the mean ± SEM. Student’s t tests were used for statistical analyses, except for (A), where survival was compared using the log-rank test. *P < 0.05. KC, keratinocyte-derived chemokine; IL-6, interleukin-6; GM-CSF, granulocyte-macrophage colony-stimulating factor.
The study shows that mycobacteria use a component of their cell wall, a branched α-glucan, to bind dectin-1, a receptor best known for recognizing fungal molecules. Rather than helping eliminate the bacteria, dectin-1 signalling unexpectedly supports mycobacterial survival. Mice lacking dectin-1 were more resistant to infection, showing lower bacterial loads, reduced inflammation, and fewer infiltrating immune cells.
At the cellular level, macrophages without dectin-1 were less able to support intracellular bacterial growth. Further analysis revealed that dectin-1 alters how phagosomes mature and interact with autophagy pathways, processes that normally help destroy invading microbes. By interfering with these pathways, mycobacteria create a niche that allows them to persist inside host cells.
Structural studies identified branched α-glucan as the specific mycobacterial ligand responsible for activating dectin-1. This interaction represents a previously unrecognized strategy by which mycobacteria manipulate innate immune signalling to evade destruction.
Together, these findings uncover a novel mechanism of intracellular immune evasion and suggest that targeting the dectin-1–α-glucan interaction could be a new approach to limiting mycobacterial survival during infection.
Journal article: Torigoe, S., et al. 2026. Mycobacterial α-glucans hijack dectin-1 to facilitate intracellular bacterial survival. Science Immunology.
Summary by Stefan Botha



