





- Patient Presentation
- History
- Differential Diagnosis
- Examination
- Investigations
- Discussion
- Treatment
- Final Outcome
- References
- Evaluation - Questions & Answers
- MCQ
Patient Presentation
A 22-year-old male presented to his physician complaining of a fever and sore throat with enlarged, tender lymph nodes in his neck for the last 24 hours. He was now feeling very ill, and his symptoms were getting worse.
History
Patient had been well preceding this acute presentation and he was not aware of any recent contact with anyone who was ill or complained of similar symptoms.
Of note was a recent travel history to a game farm in southern Africa. Malaria prophylaxis had been taken, however the patient confirmed having sustained several mosquito bites. The patient also reported finding ticks on his legs on two occasions.
Past medical history
- At age 11 he was diagnosed and treated for Non-Hodgkin’s Lymphoma (NHL). He has remained in remission.
Past surgical history
- No previous surgical history
Family History
- Nothing of significance
Allergies
- Nil known
Medication
- Nil
Travel History
- As noted above
Social history
- Non smoker
- No alcohol
- No illegal substance use
Differential Diagnosis
- Rickettsia
- Measles
- Viral hepatitis
- Malaria
- EBV
- CMV
- HSV
- Rift Valley Fever
- Typhoid
- Crimean Congo haemorrhagic fever (CCHF)
Examination on admission
Appearance:
- Ill-looking young man.
- Awake, alert and co-operative.
- Not encephalopathic
- GCS 15/15
Vitals
- Temperature: 38.5°C, febrile
- Blood pressure: 124/76
- Heart rate: 100
- Respiratory rate: 16
General
- +++ jaundice
- ++ submental lymphadenopathy, nodes soft and tender
- slight periorbital oedema
Head and Neck
- No neck stiffness
- Vision and hearing intact
- Pupils equal and reactive to light
- No papilloedema
ENT
- Palatal petechiae noted on the posterior oropharynx
- Nonexudative red inflamed pharynx
- Enlarged palatine tonsils
Chest
- Chest clear
Cardiovascular
- Normotensive
- No murmurs, no added heart sounds
Abdomen
- Mild tenderness over liver and spleen with accompanying hepatosplenomegaly.
Neurological
- Normal level of consciousness.
- Alert and co-operative.
- Sensation intact in all limbs
- Power 5/5 in all limbs
- Tone normal in all limbs
- Reflexes 2/4 in all limbs
- Gait, normal, walking unassisted.
- No seizures reported or observed.
Dermatological
- Faint, generalized maculopapular rash.
- Non pruritic.
The patient was admitted to an isolation cubicle, while awaiting blood results.
Investigations
Admission blood results
| Examination | Value | Normal Limits |
|---|---|---|
| WBC | 19.010000000000002 | (4-12 x109/L) |
| Neutrophils abs | 4.37 | (2.00-7.50 109/L) |
| Lymphocytes abs | 14.26 | (1.00-4.00 109/L) |
| Monocytes abs | 0.38 | (0.18-0.80 109/L) |
| Eosinophils abs | 0 | (0.00-0.45 109/L) |
| Basophils abs | 0 | (0.00-0.20 109/L) |
| HB | 12 | (12.1-15.2 g/L) |
| Platelets | 138 | (140-450 x109/L) |
| NA | 132 | (135-147 mmol/L) |
| K | 4.5999999999999996 | (3.3-5.0 mmol/L) |
| CL | 94 | (99-103 µmol/L) |
| C02 | 22 | (18-29 mmol/L) |
| Urea | 9.4 | (2.5-6.4 mmol/L) |
| Creatinine | 152 | (62-115 mmol/L) |
| Bilirubin total | 127 | (5-21 umol/l) |
| Bilirubin conjugated | 81 | (0-5 umol/l) |
| Alkaline phosphatase | 383 | (40-130u/L) |
| Gamma GT | 285 | ( |
| ALT | 407 | ( |
| AST | 682 | ( |
| Total Protein | 57 | (60-83 g/L) |
| Albumin | 23 | (35-52 g/L) |
| Hepatitis A IgM | Negative | |
| Hepatitis A IgG | Negative | |
| Hepatitis B sAg | Negative | |
| Hepatitis B sAb | 1.1000000000000001 | (0.0-9.9 mIU/mL) |
| Hep B sAb interpret | Negative | |
| Hepatitis B cAb | Negative | |
| Malaria thin smear | Negative | |
| Malaria thick smear | Negative | |
| P. falciparum Ag | Negative | |
| Com plasmodium Ag | Negative | |
| Measles IgM | Negative | |
| R. conorii serology | Negative | |
| HSV PCR | Negative | |
| CCHF– PCR and Serology | Negative | |
| EBV VCA IgM | Positive | |
| EBV VCA IgG | Positive | |
| EBNA IgG | Negative |
VHF serology and PCR – negative
EBV viral load on whole blood was massively elevated at log10 6.3 genome copies /mL
Over the course of the next 7 days the patient continued to deteriorate, developing metabolic acidosis, liver failure, renal failure, DIC, ARDS and neurological complications. A liver biopsy was performed and showed submassive periportal necrosis.
Discussion
Epstein-Barr virus
Background
To first understand what is happening in our patient, lets go into some background. Epstein-Barr virus (EBV) or human herpesvirus 4, is a B lymphotropic gamma-herpesvirus that infects more than 90% of the world’s population. The immune response mounted against EBV is usually quite effective in containing EBV infection, such that most individuals that have it end up being asymptomatic (Hatton et al., 2014). Other times, certain individuals will present with acute infectious mononucleosis, a self-limiting clinical syndrome that most frequently affects adolescents and young adults and is taken to be the most common manifestation of primary infection with this organism (Hatton et al., 2014). Classic symptoms include sore throat, fever and lymphadenopathy that may, or may not, be accompanied by a faint generalized maculopapular rash. Humans are the only known reservoir of EBV. In other instances, individuals can present with malignancies within lymphoid organs or the epithelium as a result of infection with EBV (Hatton et al., 2014).
To better understand the pathophysiology of EBV infection that is occurring in our case, the following series of graphics explains both the normal activation pathway of B cells when stimulated by antigen and the mechanism by which EBV drives naïve infected B cells to recapitulate this pathway.
We also explain the immunological events that occur in X-linked lymphoproliferative disease (XLP), a rare primary immunodeficiency in which sufferers have a selective inability to control EBV infection and which we speculate is the likely cause of the patient’s condition in our case study.
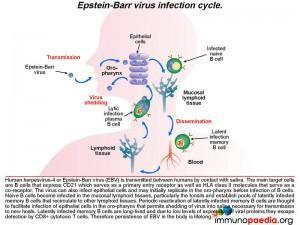 Pathophysiology of Epstein Barr virus (EBV)
Pathophysiology of Epstein Barr virus (EBV)
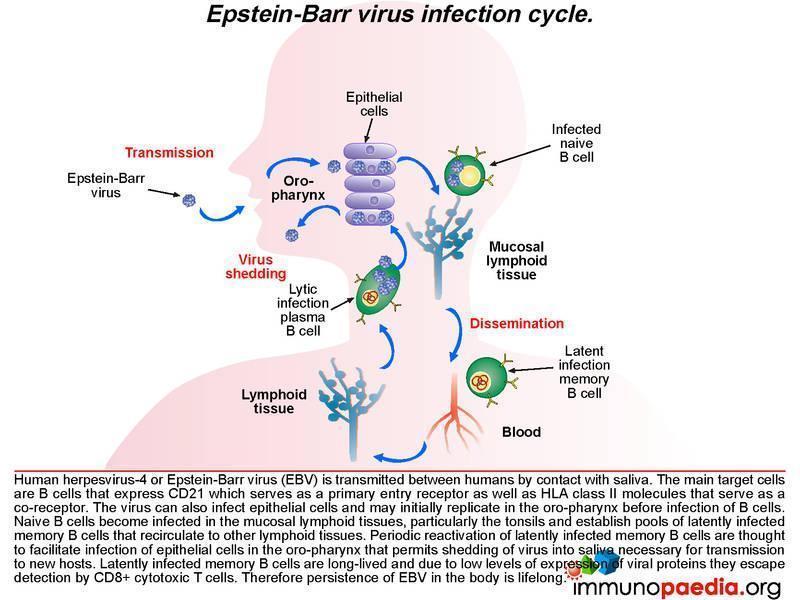
The infection life cycle of Epstein Barr virus
EBV can be transmitted through saliva. After exposure, EBV establishes a persistent infection in the host and is intermittently shed in oropharyngeal secretions. In uncomplicated EBV infections the main target cells are B lymphocytes that express CD21/ C3d which, along with HLA class II molecules, which serve as the viral entry receptor and co-receptor, respectively. The virus can also infect epithelial cells and may initially replicate in the oropharynx before infecting B cells. Naive B cells become infected in local lymphoid tissue, most often Waldeyer’s tonsillar ring, and establish latent infected memory B cell pools that migrate to other lymphoid tissues. Periodic reactivation of latently infected memory B cells are thought to facilitate infection of epithelial cells in the oropharynx that permits shedding of virus into saliva for transmission to new hosts. Latently infected memory B cells are long-lived and due to low levels of expression of viral proteins they escape detection by CD8+ cytotoxic T cells. Therefore, persistence of EBV in the body is life-long
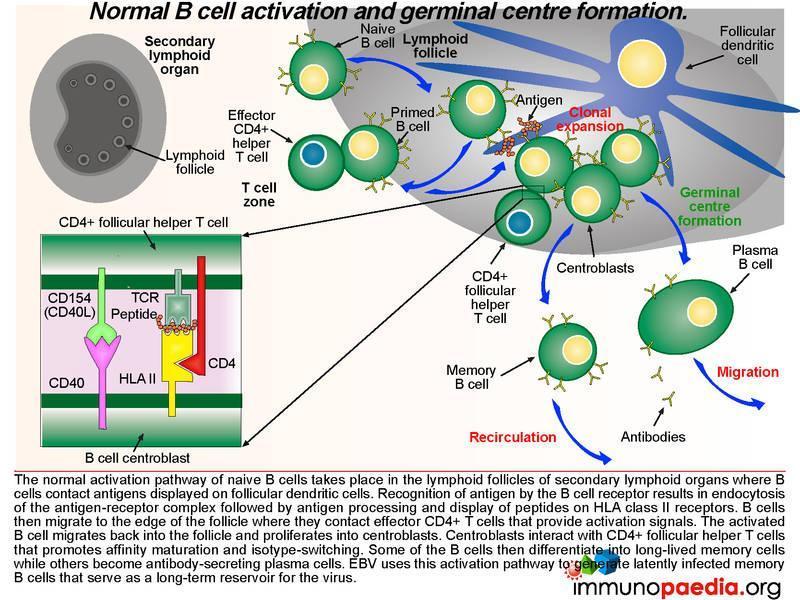 What happens in the lymph nodes
What happens in the lymph nodes
The normal activation pathway of naive B cells takes place in the lymphoid follicles of secondary lymphoid organs where B cells contact antigens displayed on follicular dendritic cells. Recognition of antigen by the B cell receptor results in endocytosis of the antigen-receptor complex followed by antigen processing and display of EBV-derived peptides on HLA class II molecules. B cells then migrate to the edge of the follicle where they contact effector CD4+ T cells that provide activation signals. The activated B cell migrates back into the follicle and proliferates into centroblasts. Centroblasts interact with CD4+ follicular helper T cells that promotes affinity maturation and isotype-switching. Some of the B cells differentiate into long-lived memory cells while others become antibody-secreting plasma cells. Epstein-Barr virus uses this activation pathway to generate latently infected long-lived memory B cells that serve as a life-long reservoir for the virus. Take note in graphic 2 of the box insert. This shows the receptor-ligand interactions that occur between the B cell centroblast and the CD4+ T cell (which is a follicular helper T cell). There are many more interactions, not shown, but the two important ones are the TCR-peptide-HLA complex and the CD40-CD40 ligand (CD154) interaction.
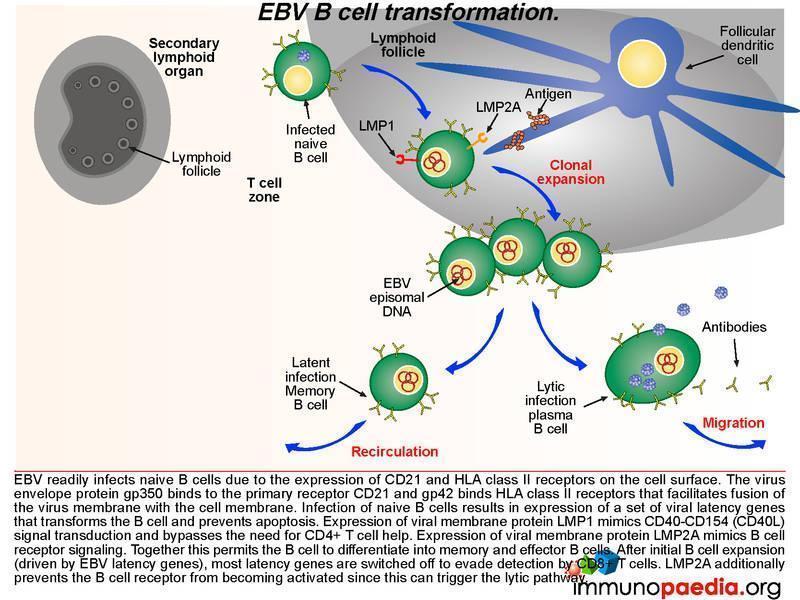 Germinal centre EBV B cell transformation
Germinal centre EBV B cell transformation
Lets look a little closer at some of the molecular events occurring in a B cell once it becomes infected with EBV. After infection of naive B cells, EBV expresses a set of latency-associated genes. The function of these genes is to immortalize the infected cell. These genes encode for two key proteins: Latent Membrane Protein-1 (LMP1) and Latent Membrane Protein 2A (LMP2A). LMP1 is a viral homologue of CD40, which allows activation of B cells without the need to engage with CD40L on the CD4+ T cell. Thus, LMP-1 expression mimics CD40-CD40L signal transduction and bypasses the need for CD4+ T cell help.
LMP2A is a B cell receptor homologue. It mimics B cell receptor signaling and coupled to LMP1 signaling promotes the B cell to undergo affinity maturation, isotype switching and differentiation into memory and effector B cells. After initial expansion, EBV-infected B cells become quiescent memory cells and viral gene expression is kept to a minimum. In fact, healthy EBV-infected individuals carry a number of these memory cells for life, and they function as a reservoir from which virus can reactivate periodically and be shed in the oropharynx.
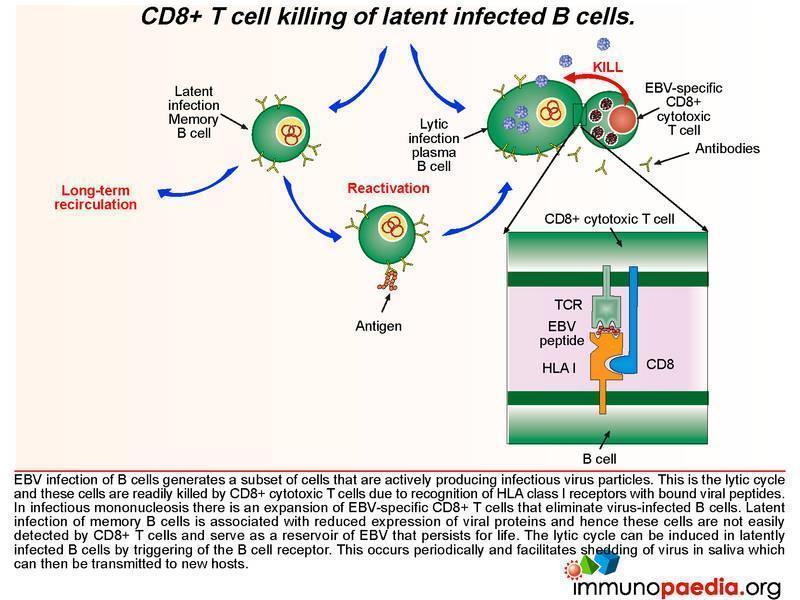 Lytic and latent pathways
Lytic and latent pathways
EBV infection of B cells generates a subset of cells that are actively producing infectious virus particles. This is the lytic cycle and these cells are readily killed by CD8+ T cells due to recognition of HLA class I receptors with bound viral peptides. This is shown in graphic 4, with the box insert. During infectious mononucleosis there is a large expansion of EBV-specific CD8+ cytotoxic T lymphocytes that eliminate virus producing B cells.
After the initial expansion of B cells, driven by LMP1 and LMP2A, latent infection of memory B cells is associated with a lack of expression of viral proteins and the ability to escape detection by CD8+ cytotoxic T lyphocytes and and hence these cells persist as a reservoir of EBV. The lytic cycle can be induced again in latently infected B cells by triggering of the B cell receptor. This occurs periodically to facilitate shedding of virus in saliva and allow transmission to new hosts.
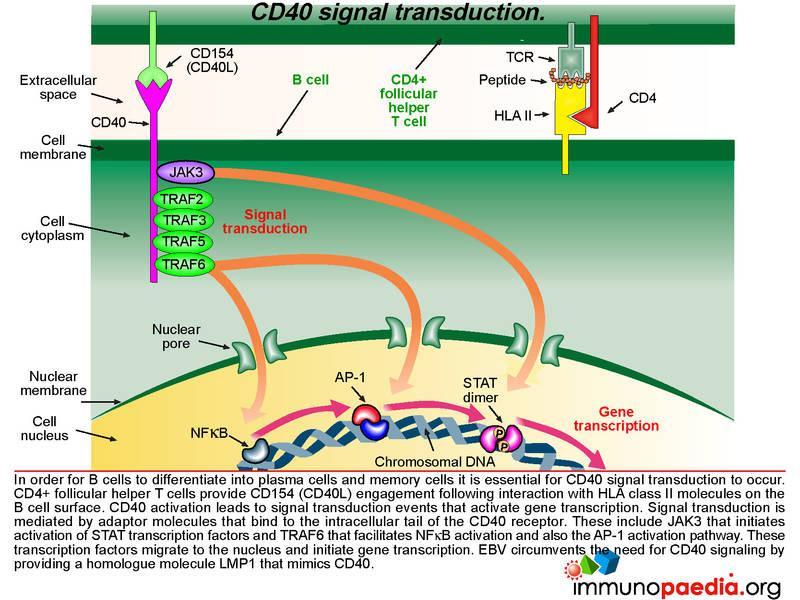 CD40 signal transduction
CD40 signal transduction
Lets look still closer at the detail of the molecular events when CD40 is activated by CD40 ligand (CD154). This is an important step that allows B cells to differentiate into plasma cells and memory cells as well as promote affinity maturation and isotype switching. CD4+ T cells provide CD154 engagement following interaction with HLA class II molecules on the B cell surface. CD40 activation leads to signal transduction events that activate gene transcription. Signal transduction is mediated by adaptor molecules that bind to the intracellular tail of the CD40 receptor. These include JAK3 that initiates the STAT transcription factor activation and TRAF6 that facilitates NFkB activation and also the AP-1 activation pathway. These transcription factors migrate to the nucleus and initiate gene transcription. EBV hijacks this process by mimicking CD40. Lets see how this is done.
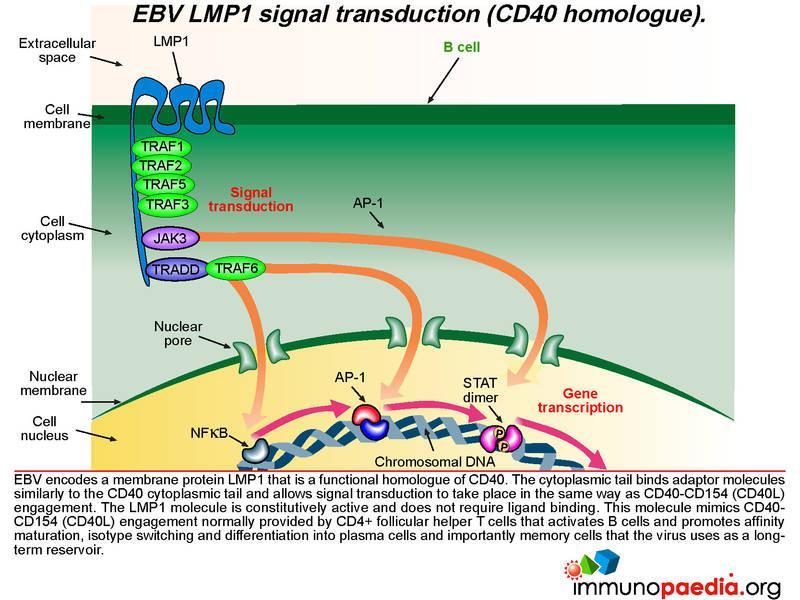 How does LMP1 serve as a CD40 homologue ?
How does LMP1 serve as a CD40 homologue ?
Epstein-Barr virus encodes a membrane protein called LMP1 that is a functional homologue of CD40. The cytoplasmic tail of LMP1 binds the same adaptor molecules as the CD40 cytoplasmic tail and allows signal transduction to take place in the same way as if CD40 engagement had occurred. The LMP1 molecule is constitutively active and does not require ligand binding. This molecule mimics the CD40-CD154 engagement normally provided by CD4+ T cells to activate B cells and allow differentiation into plasma cells and importantly memory cells that the virus uses as a reservoir.
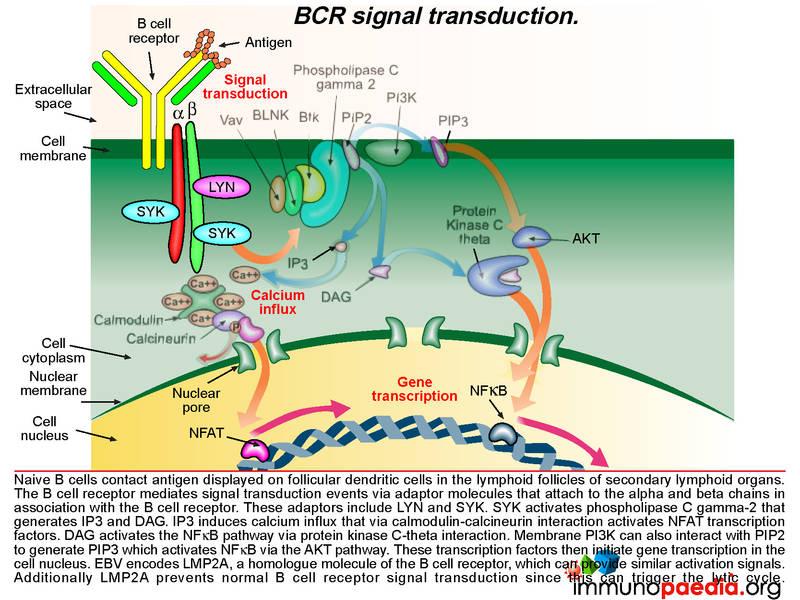 BCR signal transduction
BCR signal transduction
As already described, naive B cells interact with antigen displayed on follicular dendritic cells in the germinal centres of secondary lymphoid organs via their B cell receptors. The B cell receptor mediates signal transduction events via another set of adaptor molecules that attach to the alpha and beta chains that associate with the B cell receptor. These adaptor molecules include LYN and SYK. SYK activation leads to activation of phospholipase C gamma-2 that generates IP3 and DAG. IP3 induces calcium influx that initiates gene transcription through a series of molecular interactions. Graphic 7 shows the details of this interaction.
The important point about EBV, is that it encodes a homologue of the B cell receptor: LMP2A.
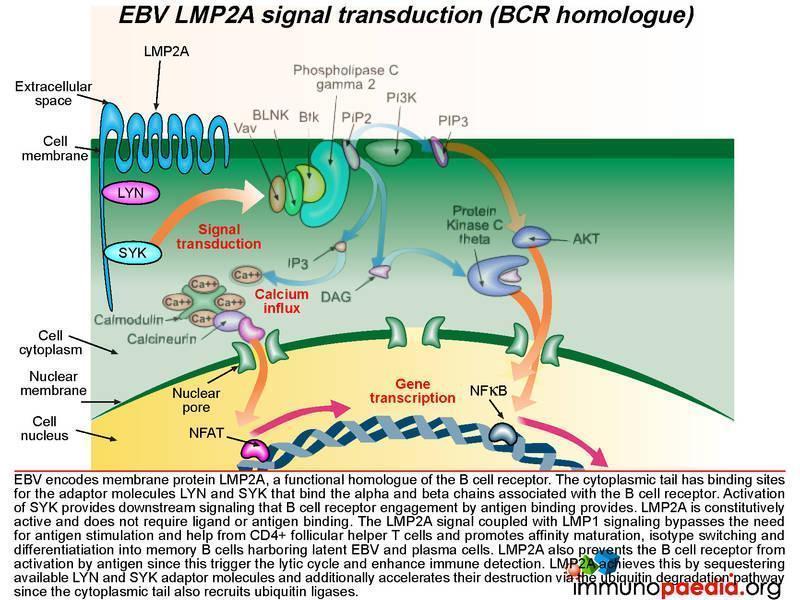 How does LMP2A serve as a B cell receptor homologue?
How does LMP2A serve as a B cell receptor homologue?
Epstein-Barr virus encodes the membrane protein LMP2A that is a functional homologue of the B cell receptor. Like LMP1, LMP2A is another viral gene during the latent phase of EBV infection that allows infected B cells move through germinal centre reactions. LMP2A’s cytoplasmic tail has binding sites for the same adaptor molecules that bind the alpha and beta molecules of the B cell receptor, including LYN and SYK. Activation of SYK provides the same down stream signaling processes that B cell receptor engagement by antigen binding provides. LMP2A is constitutively active and does not require ligand or antigen binding. The LMP2A signal coupled with LMP1 signaling thus bypasses the need for antigen stimulation and CD4+ T cell help and permits the B cell to differentiate into memory B cells containing latent EBV.
LMP2A also blocks the B cell receptor from activation by antigen because this can initiate the lytic cycle and allow detection by the immune system. LMP2A achieves this by sequestering LYN and SYK adaptor molecules and additionally accelerating their destruction via the ubiquitin degradation pathway since the cytoplasmic tails also recruit ubiquitin enzyme complexes.
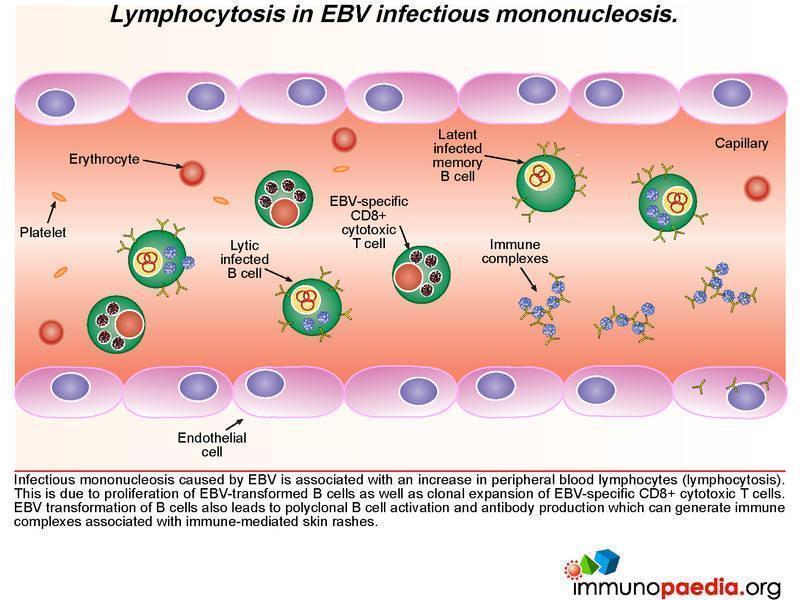 Responding CD8+ T cells, cytokine storm and clearance of EBV infected B cells
Responding CD8+ T cells, cytokine storm and clearance of EBV infected B cells
In the clinical work-up of our patient, the full blood count showed an elevated WBC and lymphocyte count indicating lymphocytosis, which possibly suggests proliferation of EBV-transformed B cells as well as clonal expansion of EBV-specific CD8+ cytotoxic T cells. EBV viraemia, which was very high in the patient, can lead to an accumulation of immune complexes, which are associated with immune mediated rashes – seen in our patient as a faint, generalized maculopapular rash.
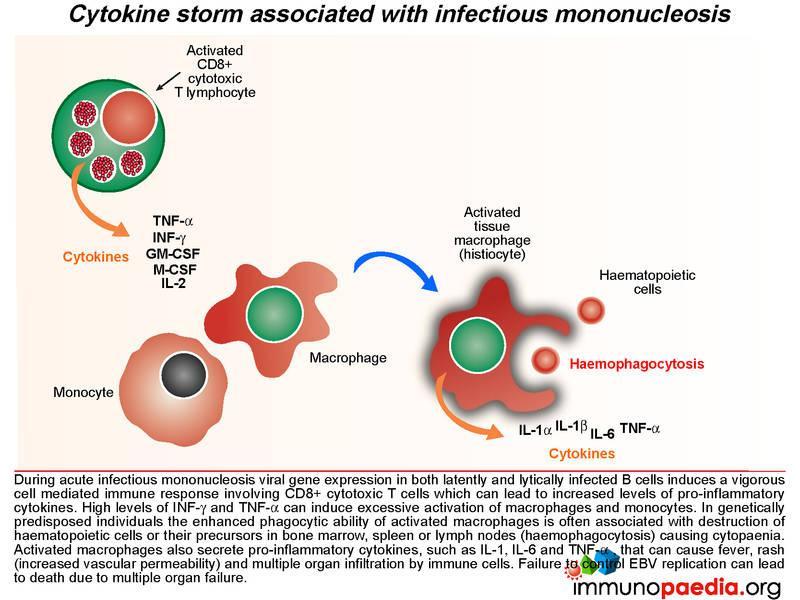 During acute infectious mononucleosis viral gene expression in both latently and lytically infected cells induces vigorous cell mediated immune responses, which can lead to increased levels of pro-inflammatory cytokines and a cytokine storm. High levels of INF-g and TNF-a can induce excessive activation of macrophages and monocytes. In genetically predisposed individuals, the enhanced phagocytic ability of activated macrophages is often associated with destruction of haematopoietic cells or their precursors in bone marrow, spleen and lymph node(haemo phagocytosis) causing cytopaenia. Activated macrophages also secrete pro-inflammatory cytokines, such as IL-1, IL-6 and TNF-a , that can cause fever, rash (increased vascular permeability) and multiple organ infiltration by immune cells. Failure to control EBV infection can lead to death due to multiple organ failure – of which was seen in this case study. (see related case)
During acute infectious mononucleosis viral gene expression in both latently and lytically infected cells induces vigorous cell mediated immune responses, which can lead to increased levels of pro-inflammatory cytokines and a cytokine storm. High levels of INF-g and TNF-a can induce excessive activation of macrophages and monocytes. In genetically predisposed individuals, the enhanced phagocytic ability of activated macrophages is often associated with destruction of haematopoietic cells or their precursors in bone marrow, spleen and lymph node(haemo phagocytosis) causing cytopaenia. Activated macrophages also secrete pro-inflammatory cytokines, such as IL-1, IL-6 and TNF-a , that can cause fever, rash (increased vascular permeability) and multiple organ infiltration by immune cells. Failure to control EBV infection can lead to death due to multiple organ failure – of which was seen in this case study. (see related case)
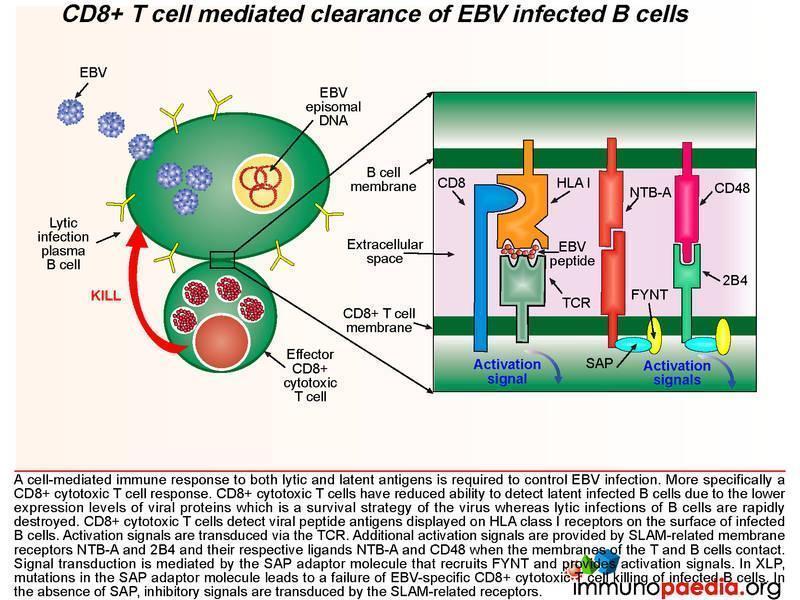 During acute infection, CD8+ T cells detect viral peptide antigens displayed on HLA class I on the surface of infected B cells. The interaction of the T cell receptor (TCR) with the EBV-derived peptide and HLA class I molecule leads to signal transduction in the CD8+ T cell. Additional activation signals are provided by signaling lymphocytic activation molecule (SLAM)-related membrane receptors NTB-A and 2B4 and their respective ligands NTB-A and CD48 when the membrane of the T and B cell make contact. Signal transduction is mediated by the SAP adaptor molecule that recruits FYNT and provides activation signals.
During acute infection, CD8+ T cells detect viral peptide antigens displayed on HLA class I on the surface of infected B cells. The interaction of the T cell receptor (TCR) with the EBV-derived peptide and HLA class I molecule leads to signal transduction in the CD8+ T cell. Additional activation signals are provided by signaling lymphocytic activation molecule (SLAM)-related membrane receptors NTB-A and 2B4 and their respective ligands NTB-A and CD48 when the membrane of the T and B cell make contact. Signal transduction is mediated by the SAP adaptor molecule that recruits FYNT and provides activation signals.
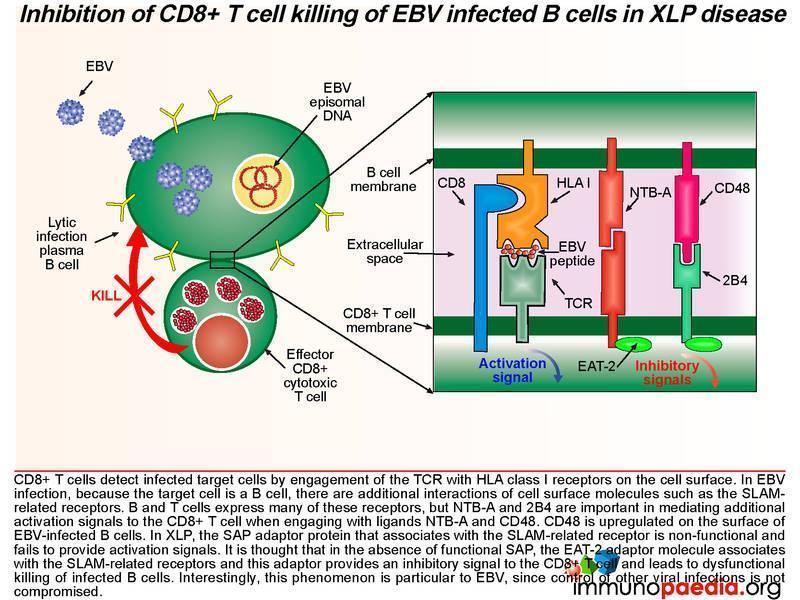
Inhibition of CD8+ T cell killing of EBV infected B cells in X-linked lymphoproliferative disease (XLP) disease.
XLP, also referred to as Duncan’s disease, affects males and is categorized as a primary immunodeficiency that results in the inability to completely clear EBV (Panchal et al., 2018). Molecular characterisation now classifies XLP as having two disease types;
- XLP type 1/XLP1: a result of mutations in the SH2D1A gene which encodes the cytoplasmic adaptor protein Signaling Lymphocytic Activation Molecule (SLAM)-associated protein (SAP). Males present with dysgammaglobulinemia, disseminated lymphoproliferative disorder, aplastic anemia, vasculitis, chronic gastritis, and skin lesions (Panchal et al., 2018).
- XLP type 2/XLP2: a result of mutations in the BIRC4 gene, that encodes for XIAP, the X-linked inhibitor of apoptosis. The major presentation of this disease is hemophagocytic lymphohistiocytosis (HLH) as well as splenomegaly and colitis which are not consistently seen in males with XLP1 (Zhang, Wakefield and Marsh, 2016).
XLP1 taken to be more common than XLP2 and estimated to affect about 1 per million males across the globe (Seemayer, 1993).
SAP is a SLAM associated protein that contains a Src homology 2 (SH2) domain and is expressed on T cells, NK cells, and some EBV-positive Burkitt lymphoma-derived B cells. SAP bind to SLAMs through the SH2 domain, contributing to the development of invariant natural killer T cells (iNKT) and the killing of EBV-infected B cells through cytotoxic T lymphocytes (CTLs) and NK cells (Wang et al., 2022) . XLP1 leads to a deficiency of SAP thus blocking cytotoxic T cell responses to EBV-infected B cells. CD8+ T cells detect infected target cells by engagement of the TCR with HLA class I receptors on the cell surface. In EBV infection, because the target cell is a B cell, there are additional interactions of cell surface molecules such as the SLAM related receptors. B and T cells express many of these receptors, but NTB-A and 2B4 are important in mediating additional activation signals to the T cell when engaging with ligands NTB-A and CD48. Also, CD48 is upregulated on the surface of EBV-infected B cells. In XLP, the SAP adaptor protein that associates with the SLAM-related receptor is non-functional and fails to provide activation signals. It is thought that in the absence of SAP, the EAT-2 adaptor molecule associates with the SLAM-related receptors and this adaptor provides an inhibitory signal to the T cell and leads to dysfunctional CD8+ T cell killing of infected B cells.
Thus EBV infected B cells are not killed in the acute stage of infection. Virus replication and B cell proliferation then continues unchecked and multi-organ failure can occur following primary infection as a result of the ongoing cytokine storm.
Download images for this case
Treatment
The patient was treated initially with corticosteroids to decrease the swelling in his throat and to decrease the splenomegaly and prevent possible splenic rupture.
Due to his continued disease progression he was switched to the HLH-94 (haemophagocytic lymphohistiocytosis) protocol of steroids and cyclosporine which is recommended for patients who develop fulminant disease. However the patient failed to respond to this treatment regimen.
Additionally he was given IVIG and a trial of Rituximab (anti-CD20) but he did not respond to either therapy.
Download images for this case
Final Outcome
The patient rapidly deteriorated, failing all treatment. He developed multi organ failure, and died one week after admission.
Although no genetic testing was done on this patient and given that he was male, we speculate that this was an X-linked lymphoproliferative disease. Thus, genetic testing should be offered to his family as it is important to identify other affected males and female carriers.
For affected males allogenic stem cell transplant performed prior to EBV exposure is curative.
Female carriers can be offered prenatal diagnosis and their male infants can be screened at birth – which would allow for allogenic stem cell transplants to be performed. A definitive diagnosis of XLP is genetic screening of the SH2D1A gene that encodes the SAP adaptor molecule.
Download images for this case
References
Okano M, Gross TG. (2011) Acute or Chronic Life-Threatening Diseases Associated With Epstein-Barr Virus Infection. Am J Med Sci.Nov 17. [Epub ahead of print]
Palendira U et al. (2011) Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol. Nov;9 (11):e1001187
Chaganti S, Ma CS, Bell AI, et al. (2008) Epstein-Barr virus persistence in the absence of conventional memory B cells: IgM+IgD+CD27+ B cells harbor the virus in X-linked lymphoproliferative disease patients. Blood. Aug 1;112(3):672-9
Hurt C, Tammaro D. (2007) Diagnostic evaluation of mononucleosis-like illnesses. Am J Med. Oct;120(10):911.e1-8
Luzuriaga K, Sullivan JL. (2010) Infectious mononucleosis. N Engl J Med. May 27;362(21).
Hatton, O. L. et al. (2014) The interplay between Epstein-Barr virus and B lymphocytes: implications for infection, immunity, and disease. Immunologic research, 58(2–3), pp. 268–276.
Panchal, N. et al. (2018) X-linked lymphoproliferative disease type 1: A clinical and molecular perspective. Frontiers in immunology, 9.
Zhang, K., Wakefield, E. and Marsh, R. (2016) Lymphoproliferative Disease, X-Linked. University of Washington, Seattle.
Seemayer, T. A. (1993) “X-linked Lymphoproliferative Disease,” Archives of pediatrics & adolescent medicine, 147(11), p. 1242.
Wang, Yanchun et al. (2022) “Potential pathogenic mechanism of type 1 X-linked lymphoproliferative syndrome caused by a mutation of SH2D1A gene in an infant: A case report,” Medicine, 101(41), p. e30951.
Download images for this case
Evaluation – Questions & Answers
What was the final diagnosis?
The likely cause for the clinical presentation was acute infectious mononucleosis and the fulminant clinical course raises the possibility that he could have had X-linked lymphoproliferative disorder. The following facts support this diagnosis:
- Male sex (XLP is a X-linked recessive condition)
- No previous history of immunodeficiency towards any other organism.
- Past history of NHL. B cell lymphomas commonly occur in the patients with this disorder.
However, no genetic testing was done to confirm the clinical suspicion.
In uncomplicated EBV infections which are the main target cells for infection?
Following antigen presentation and peptide processing, B cells then require CD4+T cell help, for what process?
How does EBV immortalize B cells?
How does EBV manage to persist life long after infection?
How is EBV infection controlled?
A cell-mediated immune response by CD8+ cytotoxic T cells directed
towards lytic and latent EBV peptides presented on infected B cells. The CD8+ T cells detect viral peptide antigens displayed on HLA class I receptors on the surface of infected B cells. Activation signals are transduced via the TCR and by SLAM-related membrane receptors NTB-A and 2B4 and their respective ligands NTB-A and CD48
Download images for this case
Multiple Choice Questions
Earn 1 HPCSA or 0.25 SACNASP CPD Points – Online Quiz
Download images for this case



