





- Patient Presentation
- History
- Differential Diagnosis
- Examination
- Investigations
- Discussion
- Diagnosis
- Treatment
- References
- Evaluation - Questions & answers
- MCQ
Patient Presentation
Mr GH was a 59 year old factory worker from Cape Town. He presented to GSH with a 2 week history of fever, rigors, weakness and a fine maculopapular rash. He was also noted to have an ulcerating papule on his left forearm. He had previously been well and there was no past medical history of note. He also gave no history of recent travel outside of Cape Town. He had not been walking in the veld and did not have any pets.
On examination, he was an ill looking man, febrile (temperature of 40 oC). The only abnormal findings were the ulcerating papule on forearm, a fine macular rash, a 3cm hepatomegaly and features suggestive of pneumonitis.
Past Medical History
- Nil of note
Past Surgical History
- Nil of note
Family and Social History
- Employed factory worker
- No habits reported
Travel History
- No recent travel history noted
Initial Laboratory Findings
- Hb 11.3 g/dl, WBC 11.3 x109/L, (platelet count invalid due to clumping)
- HIV serology negative
- Sputum auramine negative
- Malaria thick and thin film negative
- Rickettsia serology (was pending at this stage)
Differential Diagnosis
- Tick bite fever
- Community Acquired Pneumonia
- Epstein-Barr Virus
- Haemophagocytic Syndrome
- Systemic Lupus Erythematosus
Initial Management:
IV antibiotics were administered, namely Ceftriaxone 1g IV od, doxycycline 100mg bd
Ongoing Clinical Course:
There was no improvement in his clinical condition while in the ward. He continued to have swinging fevers and he developed a pancytopenia. Also rickettsia serology came back negative. At this point he was referred to infectious diseases for further work up.
Examination
- Persistent fever
- Rash
- Pancytopaenia
- Hepatomegaly
- Pneumonitis
- Eschar
Investigations
| WBC | 2.2000000000000002 |
|---|---|
| Hb | 10.3 |
| PLT | 27 |
| Neut | 1.45 |
| Lymph | 0.59 |
| Fibrinogen | 4 |
| CRP | 217 |
| Coxiella Phase I & II | IgM negative |
| IgG titre 1024 | |
| Mycoplasma serology | IFA IgM negative |
| Autoimmune screen | Negative |
| Blood cultures | Negative |
| RPR | Negative |
| Legionella urinary Ag | Negative |
Bone marrow biopsy was requested:
- Hypercellular marrow.
- Poorly formed granulomas (AFB negative)
- Infiltration of plasma cells and histiocytes (CD68 pos)
- Evidence of haemophagocytosis (of granulocyte and erythroid precursors)
Assessment:
Haemophagocytic syndrome secondary to probable infection, possibly TB.
Further investigations:
Despite negative sputum smears, a CT scan of chest: was performed to look for TB. A spiculated mass was detected in Rt hilum.
Bronchoscopy and BAL brushings:
- AFB positive
- PCR positive for MTB, sensitive to rifampicin and INH
- TB culture was positive after 19 days.
- (TB culture of initial sputum was positive after 32 days)
Discussion
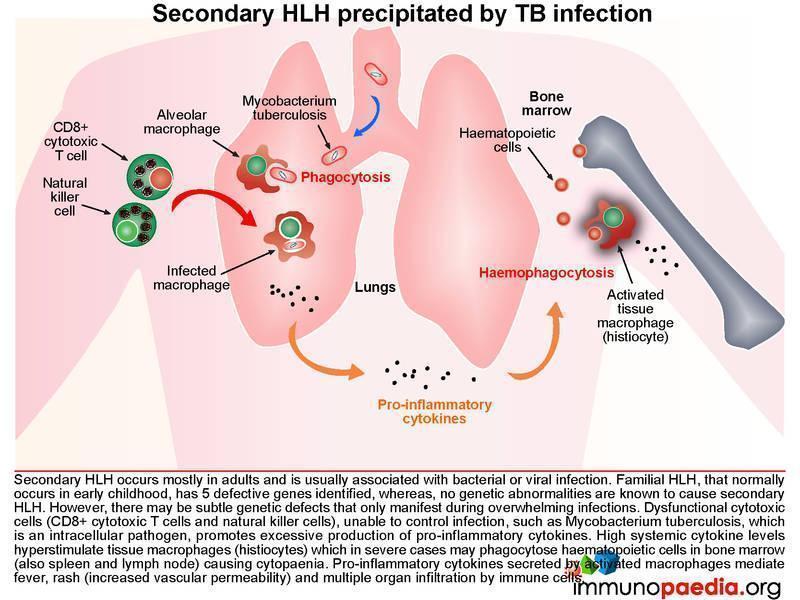 Haemophagocytic lymphohistiocytosis (HLH) is a multi-system inflammatory disorder caused by prolonged and excessive activation of immune cells, in particular natural killer (NK) cells and CD8+ cytotoxic T cells (CTL) and antigen-presenting cells. It arises when there is an ineffective cell-mediated immune response to an immune stimulus or failure of regulatory pathways responsible for contraction or termination of the specific immune response. Common triggers for HLH include infections (by intracellular pathogens), malignancies, autoimmune disease or metabolic conditions. In this case the inciting immune stimulus is believed to have been pulmonary Tuberculosis. In such a scenario, dysfunctional CTL and NK cells are believed to be ineffective in controlling the infection. As a result of underlying genetic defects the cytotoxic function of these cells is compromised. This leads to overproduction of pro-inflammatory cytokines by CTL and NK cells, which in turn hyperstimulates tissue macrophages (histiocytes). The hypercytokinaemia is responsible for the cardinal clinical features of this syndrome many of which were present in our patient, namely persistent, hectic fever, hepatosplenomegaly, seizures, ataxia, rash, acute respiratory distress syndrome (ARDS) and progressive organ dysfunction. The cytopaenias are caused by widespread phagocytosis of haematopoietic precursors by infiltrating histiocytes in the bone marrow (haemophagocytosis). Importantly, haemophagocytosis may not always be obvious on the marrow biopsy, especially early during the clinical course. This was the case in our patient. Untreated the disease is usually fatal.
Haemophagocytic lymphohistiocytosis (HLH) is a multi-system inflammatory disorder caused by prolonged and excessive activation of immune cells, in particular natural killer (NK) cells and CD8+ cytotoxic T cells (CTL) and antigen-presenting cells. It arises when there is an ineffective cell-mediated immune response to an immune stimulus or failure of regulatory pathways responsible for contraction or termination of the specific immune response. Common triggers for HLH include infections (by intracellular pathogens), malignancies, autoimmune disease or metabolic conditions. In this case the inciting immune stimulus is believed to have been pulmonary Tuberculosis. In such a scenario, dysfunctional CTL and NK cells are believed to be ineffective in controlling the infection. As a result of underlying genetic defects the cytotoxic function of these cells is compromised. This leads to overproduction of pro-inflammatory cytokines by CTL and NK cells, which in turn hyperstimulates tissue macrophages (histiocytes). The hypercytokinaemia is responsible for the cardinal clinical features of this syndrome many of which were present in our patient, namely persistent, hectic fever, hepatosplenomegaly, seizures, ataxia, rash, acute respiratory distress syndrome (ARDS) and progressive organ dysfunction. The cytopaenias are caused by widespread phagocytosis of haematopoietic precursors by infiltrating histiocytes in the bone marrow (haemophagocytosis). Importantly, haemophagocytosis may not always be obvious on the marrow biopsy, especially early during the clinical course. This was the case in our patient. Untreated the disease is usually fatal.
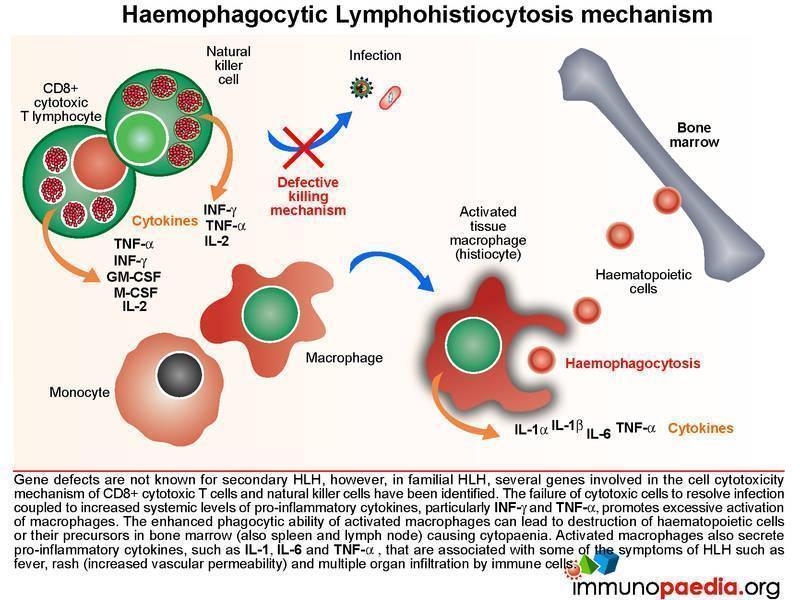 The Mechanism of Haemophagocytic Lymphohistiocytosis
The Mechanism of Haemophagocytic Lymphohistiocytosis
Both hereditary and secondary forms of this disease are recognised, but the distinction between the two has lately become blurred as it has become clear that genetic forms can present quite late in life. It may be possible that all so-called secondary forms of this condition are due to underlying (undetected) genetic disorders. In familial HLH, a growing number of autosomal recessive defects have been identified in genes that encode proteins involved in cell cytotoxic killing function (NK cells and CTLs) and activation-induced cell death (necessary for contraction of the immune response following antigen clearance). These include defects in perforin formation/function and also in key proteins required in the degranulation process following CTL/NK cell activation.
The failure of cytotoxic cells to resolve infection coupled to ongoing production of pro-inflammatory cytokines, particularly gamma-interferon (IFN-g) and tumour necrosis factor-alpha (TNF-a), mediates the hyperstimulation of macrophages. In severe cases, the enhanced phagocytic activity of these cells leads to destruction of haematopoeitic cells or their precursors in bone marrow (also observed in spleen and lymph node) that results in cytopaenia. In addition, increased synthesis of pro-inflammatory cytokines by hyperstimulated macrophages, such as IL-1, IL-6, TNF-a and IL-8 is responsible for some of the clinical symptoms such as fever, rash (increased vascular permeability) and multiple organ dysfunction due to infiltration of immune cells.
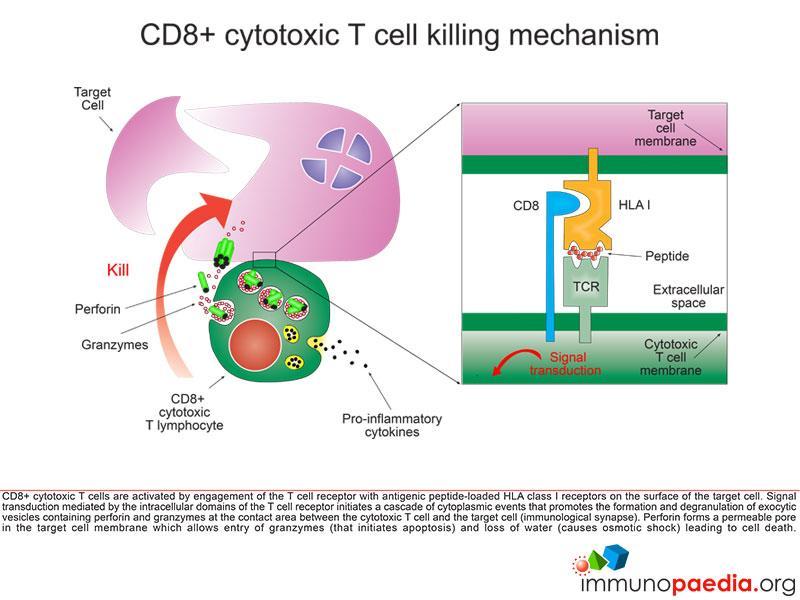 Let us look at CD8+ cytotoxic T cells in more detail
Let us look at CD8+ cytotoxic T cells in more detail
CD8+ cytotoxic T cells (CTL) become activated by engagement of the T cell receptor with antigenic peptide loaded onto HLA class I molecules on the surface of the target cell (in this case a macrophage infected with Mycobacterium tuberculosis). This receptor-ligand engagement results in intracellular signal transduction that is mediated by the intracellular domains of the T cell receptor. This promotes a cascade of events in the cytoplasm of the CTL that facilitates the formation and degranulation of exocytic vesicles containing perforin and granzymes into the intracellular space between the CTL and target cell (which we call the immunological synapse). Perforin forms a permeable pore in the target cell membrane, which allows entry of granzymes (that initiates apoptosis) and the loss of water (that causes osmotic shock), leading to cell death.
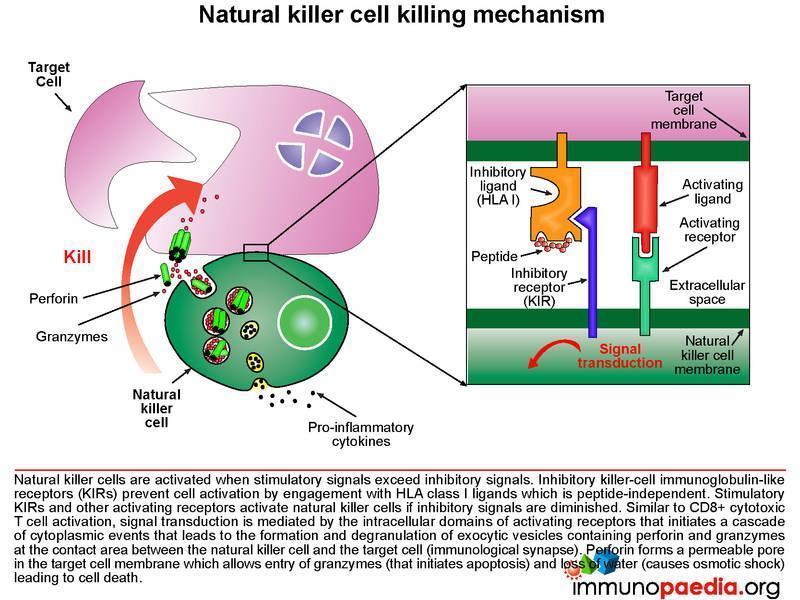 Let us look at natural killer cells in more detail
Let us look at natural killer cells in more detail
Natural killer (NK) cells form the initial defense against intracellular pathogens. NK cells do not recognise specific antigens, rather they detect “an absence of self”. That is, they recognise a potential target cell (such as virus-infected cells) by the fact that surface self HLA class I molecules are absent or diminished. Many intracellular pathogens down regulate expression of HLA class I receptors on the surface of the cells they infect in order to evade recognition by virus-specific CTLs. However, this renders them susceptible to attack by NK cells. Inhibitory killer-cell immunoglobulin-like receptors (KIRs) on the surface of the NK cell inhibit activation when they engage self HLA class I molecules. If inhibitory signals are diminished or absent, stimulatory KIRs and other activating receptors will cause NK cell activation and initiate an attack on the target cell. As with the T cell receptor, activation of the NK cell occurs following signal transduction mediated by intracellular domains when the activating receptors are triggered, which in turn promotes a cascade of events in the cytoplasm of the NK cell that leads to the formation and degranulation of exocytic vesicles containing perforin and granzymes that are released into the immunological synapse. Perforin forms a permeable pore in the cell membrane of the target cell, which allows entry of granzymes (that initiates apoptosis) and the loss of water (that causes osmotic shock) leading to cell death.
Defective cell cytotoxicity is the basis of HLH
We explain in a series of easy-to-understand graphics, the events leading to the release of lytic granules (degranulation) of both CTL’s and NK cells. By understanding these events, it is then possible to appreciate the defective proteins that interrupt these events.
The first event necessary to initiate the degranulation process of cytotoxic cells, such as CTL and NK cells, is cell activation mediated by receptor-ligand signal transduction.
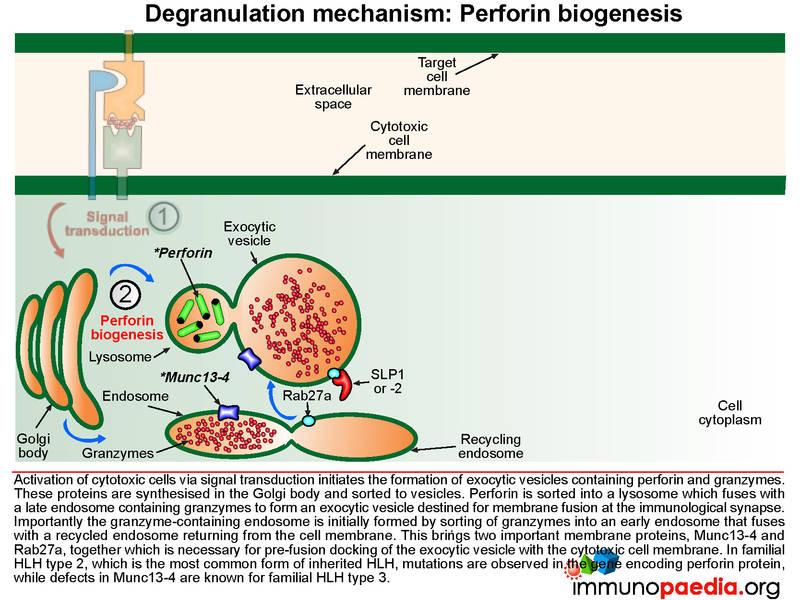
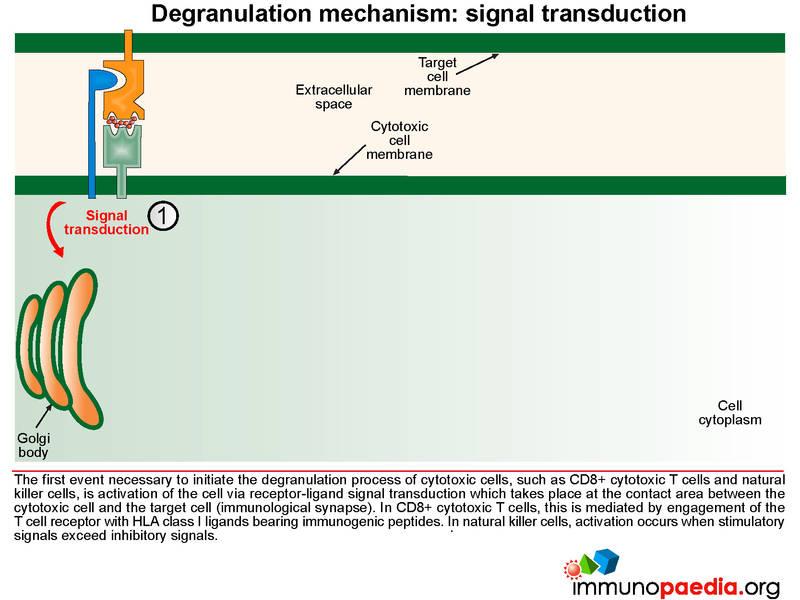 Signal transduction initiates the formation of exocytic vesicles containing perforin and granzymes. These proteins are synthesised in the Golgi body and are sorted into vesicles. Perforin is sorted into a lysosome which fuses with a late endosome containing granzymes to form an exocytic vesicle destined for membrane fusion. Importantly the granzyme-containing endosome is first formed by sorting of granzymes into an early endosome that fuses with recycling endosomes returning from the cell membrane. This brings important proteins such as Munc13-4 and Rab27a together which are required for later membrane docking. In familial HLH type 2, the most common mutations observed are in the perforin gene while mutations in Munc13-4 are known for familial HLH type 3.
Signal transduction initiates the formation of exocytic vesicles containing perforin and granzymes. These proteins are synthesised in the Golgi body and are sorted into vesicles. Perforin is sorted into a lysosome which fuses with a late endosome containing granzymes to form an exocytic vesicle destined for membrane fusion. Importantly the granzyme-containing endosome is first formed by sorting of granzymes into an early endosome that fuses with recycling endosomes returning from the cell membrane. This brings important proteins such as Munc13-4 and Rab27a together which are required for later membrane docking. In familial HLH type 2, the most common mutations observed are in the perforin gene while mutations in Munc13-4 are known for familial HLH type 3.
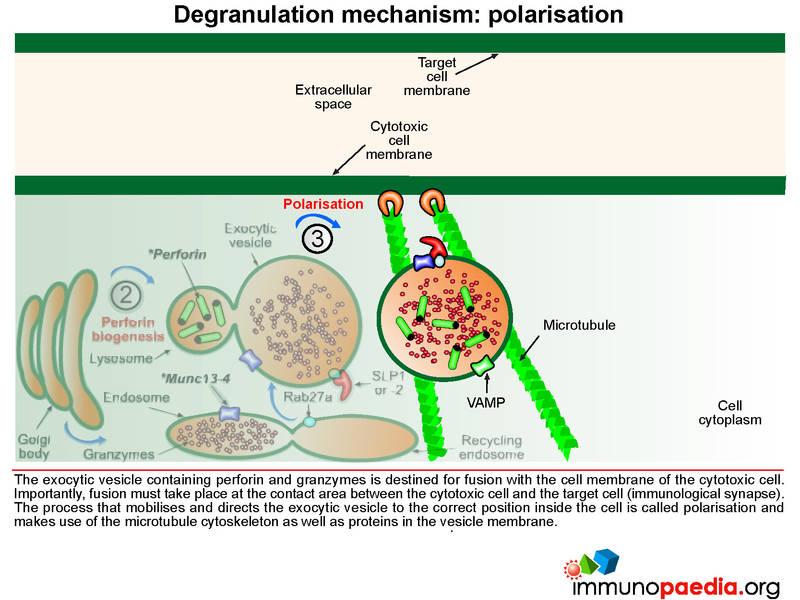
 The exocytic vesicle containing perforin and granzymes is destined for fusion with the cell membrane of the cytotoxic cell. Fusion must take place at the contact area between the cytotoxic cell and the target cell (at the immunological synapse). The process that directs the exocytic vesicle to the correct position inside the cell is called polarisation and makes use of the microtubule cytoskeleton and important proteins in the vesicle membrane.
The exocytic vesicle containing perforin and granzymes is destined for fusion with the cell membrane of the cytotoxic cell. Fusion must take place at the contact area between the cytotoxic cell and the target cell (at the immunological synapse). The process that directs the exocytic vesicle to the correct position inside the cell is called polarisation and makes use of the microtubule cytoskeleton and important proteins in the vesicle membrane.
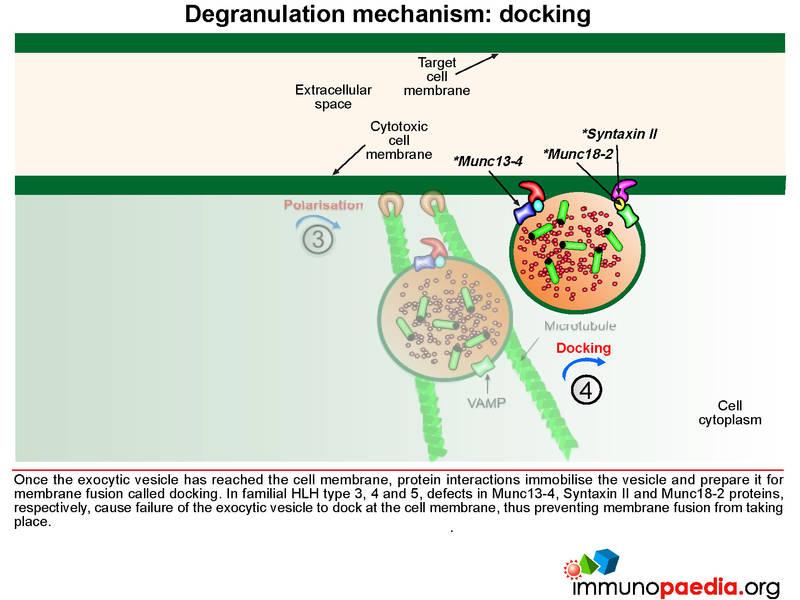
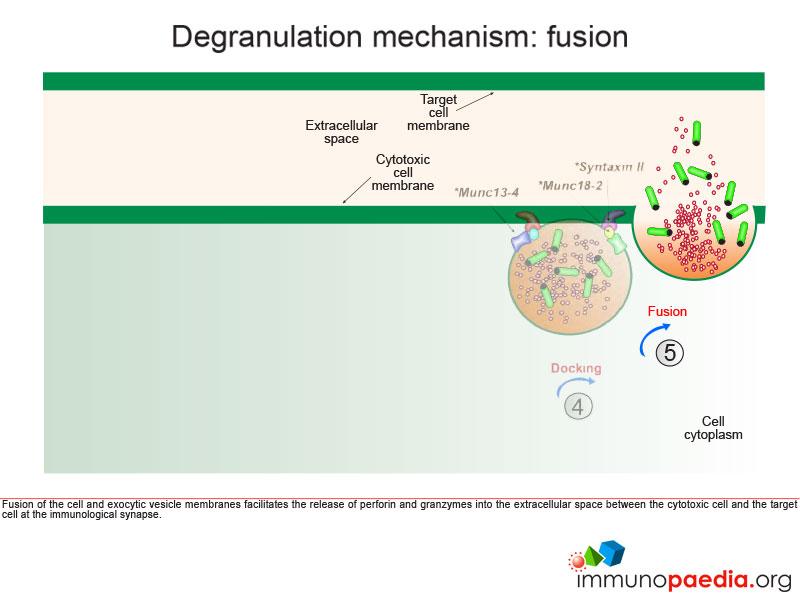
 Once the exocytic vesicle has reached the cell membrane, a process called docking takes place which involves proteins in the vesicle membrane and cell membrane that immobilise the vesicle and prepare it for membrane fusion. In HLH type 4 and 5, mutations in Syntaxin II and Munc 18-2, respectively, cause failure of the exocytic vesicle to dock at the cell membrane.
Once the exocytic vesicle has reached the cell membrane, a process called docking takes place which involves proteins in the vesicle membrane and cell membrane that immobilise the vesicle and prepare it for membrane fusion. In HLH type 4 and 5, mutations in Syntaxin II and Munc 18-2, respectively, cause failure of the exocytic vesicle to dock at the cell membrane.
Fusion of the cell membrane with the exocytic vesicle membrane releases the contents (perforin and granzymes) into the extracellular space between the cytotoxic cell and the target cell at the immunological synapse.
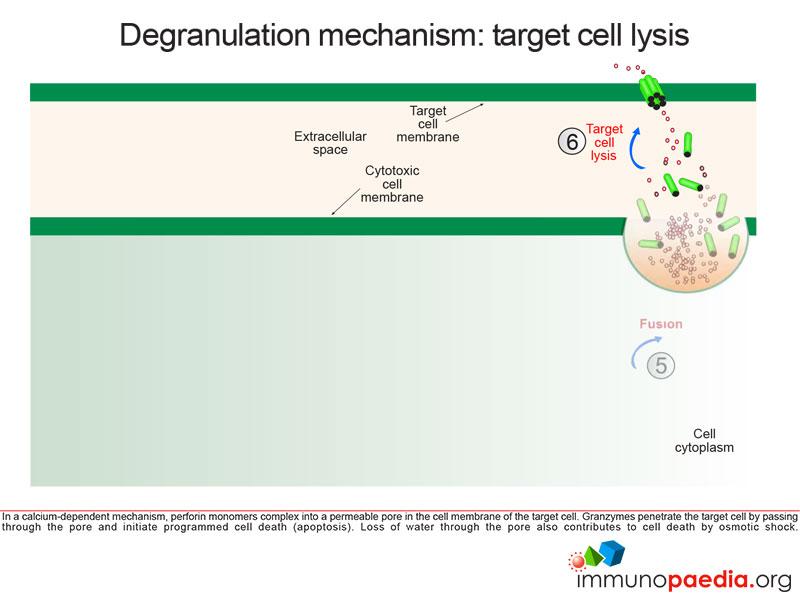
 Perforin monomers complex into a permeable pore in the cell membrane of the target cell. Granzymes penetrate the target cell by passing through the pore and initiate programmed cell death (apoptosis). Loss of water through the pore also contributes to cell death by osmotic shock.
Perforin monomers complex into a permeable pore in the cell membrane of the target cell. Granzymes penetrate the target cell by passing through the pore and initiate programmed cell death (apoptosis). Loss of water through the pore also contributes to cell death by osmotic shock.
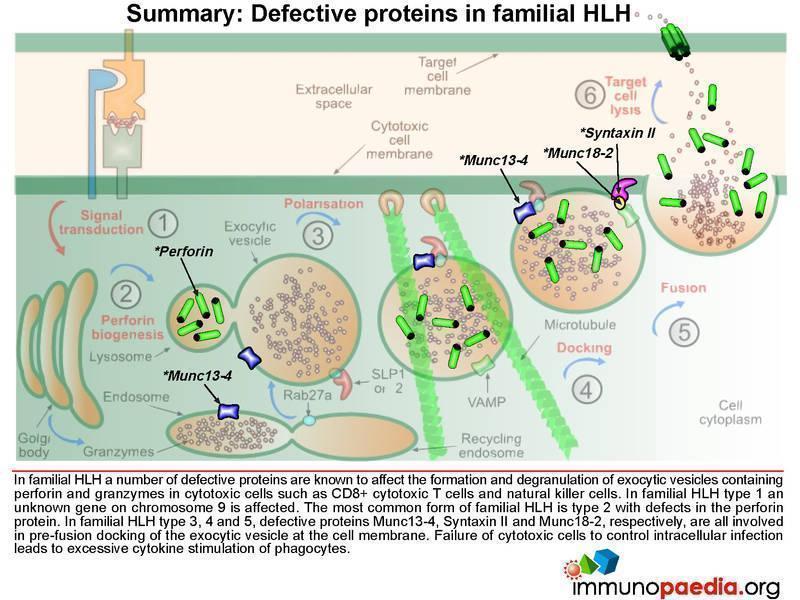
 In familial HLH a number of genetic defects are known which affect proteins involved in the process of degranulation. Five types of familial HLH have been defined (types 1-5), each associated with a specific gene defect, but additional forms are likely to be described as new gene defects are discovered. (So far all the gene defects that have been defined are autosomal recessive.) In addition HLH syndrome can complicate some other established severe-combined immunodeficiency (SCID) syndromes (see below). In familial HLH type 1 an unknown gene on chromosome 9 is affected. Perforin defects are the most common mutations observed for type 2. In types 3, 4 and 5, respective proteins Munc13-4, Syntaxin II and Munc18-2, which are involved in pre-fusion docking of the exocytic vesicle, are known to be defective.
In familial HLH a number of genetic defects are known which affect proteins involved in the process of degranulation. Five types of familial HLH have been defined (types 1-5), each associated with a specific gene defect, but additional forms are likely to be described as new gene defects are discovered. (So far all the gene defects that have been defined are autosomal recessive.) In addition HLH syndrome can complicate some other established severe-combined immunodeficiency (SCID) syndromes (see below). In familial HLH type 1 an unknown gene on chromosome 9 is affected. Perforin defects are the most common mutations observed for type 2. In types 3, 4 and 5, respective proteins Munc13-4, Syntaxin II and Munc18-2, which are involved in pre-fusion docking of the exocytic vesicle, are known to be defective.
 Active pulmonary TB is likely to have been the immunological trigger in this case. However, why he was predisposed to respond in this way is not known. No genetic studies were done to determine whether there was an underlying genetic defect to account for the HLH. The absence of previous evidence of hyperimmune reactivity suggests that any genetic defect, if present was subtle. However, it is now recognised that familial forms of HLH can present for the first time in older age (even in the 7th decade of life). It would be interesting to find out whether there was any consanguinity in the family.
Active pulmonary TB is likely to have been the immunological trigger in this case. However, why he was predisposed to respond in this way is not known. No genetic studies were done to determine whether there was an underlying genetic defect to account for the HLH. The absence of previous evidence of hyperimmune reactivity suggests that any genetic defect, if present was subtle. However, it is now recognised that familial forms of HLH can present for the first time in older age (even in the 7th decade of life). It would be interesting to find out whether there was any consanguinity in the family.
 Other protein defects in the degranulation process are known
Other protein defects in the degranulation process are known
Interestingly, haemophagocytic syndrome can also complicate a number of other defined primary immunodeficiency syndromes in which genetic defects also map to proteins important in the degranulation process: In Hermansky-Pudlak syndrome type II mutations in AP3B1 affects sorting of proteins to endosomes and also polarisation of the exocytic vesicle to the cell membrane. Similarly, Wiskott-Aldrich syndrome affects the WASp protein that is linked to cytoskeleton formation and affects polarisation of exocytic vesicles. Griscelli syndrome type 2 has defects in Rab27a that affects vesicle docking and Griscelli syndrome type 1 has defects in MyoVa which affects polarisation and migration of vesicles. Chediak-Higashi syndrome affects sorting of proteins to endosomes because of defects in LYST protein.
Download images for this case
Diagnosis
TB-associated Haemophagocytic Syndrome
Download images for this case
Treatment
Once the diagnosis of pulmonary TB was confirmed, our patient was started on TB therapy to which he responded well and was discharged 10 weeks after his initial admission.
However, 3 months on he still has compromised bone marrow function: WCC 5.86 (normal), RCC 2.44 (low), Hb 6.4 (low), platelets 39 (low).
Download images for this case
References
de Saint Basile G et al. (2010). Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. Aug;10(8):568-79.
Tang YM, Xu XJ. (2011). Advances in hemophagocytic lymphohistiocytosis: pathogenesis, early diagnosis/differential diagnosis, and treatment. ScientificWorldJournal. Mar 22;11:697-708.
Filipovich AH. (2009). Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program. 127-31.
Freeman HR, Ramanan AV. (2011). Review of haemophagocytic lymphohistiocytosis., Arch Dis Child. Jul;96(7):688-93. Epub 2010 Jun 28.
Download images for this case
Evaluation – Questions & answers
What is the diagnosis?
What are the two types of HLA?
What is the Mechanism of Haemophagocytic Lymphohistiocytosis?
How does HLH present clinically?
Respiratory and neurologic symptoms such as cough, irritability, seizures, cranial nerve palsies and ataxia have also been reported.
What is the cause of the most commonly encountered symptoms?
Download images for this case
Multiple Choice Questions
Earn 1 HPCSA or 0.25 SACNASP CPD Points – Online Quiz
Download images for this case



