





What Is Inflammation?
- Inflammation (Latin, inflammatio, to set on fire) is the complex biological response of vascular tissues to injury or harmful stimuli, such as those caused by pathogens, damaged cells, or irritants.
- In general, the following events occur during an inflammatory response:
- (1) vascular changes: blood flow increases and fluid and plasma proteins leak into the inflamed tissue;
- (2) cellular infiltration: leukocytes adhere to vascular endothelium and migrate through the endothelial layer to gain access to surrounding tissue; and
- (3) chemotaxis: leukocytes follow a chemical gradient to the site of insult and unleash potent killing mechanisms.
- Inflammation can be classified as either acute or chronic.
- Acute inflammation takes place over minutes to days and histologically is characterized by accumulation of neutrophils.
- Acute inflammation may either lead to resolution (healing), or progress to chronic inflammation.
- Chronic inflammation continues for more than a few days and is usually associated with an influx of monocytes, lymphocytes, and other immune cells.
- Chronic inflammation can represent either a progression from acute inflammation, with no resolution phase, or may also arise from a mild acute response or as a repeated low-level inflammation where no resolution phase is initiated.
- In either case, it is the persistence and failure of elimination of the foreign invader that drives the immune response to chronic inflammation.
Where Does Inflammation Fit into the Spectrum of Innate and Acquired Immune Responses?
- Inflammation is the fundamental mechanism by which multicellular organisms deal with injury caused either by infection-related or non-infection-related insults.
- Inflammation plays a central role not only in infectious diseases but also in non-infectious disease insults.
- The inflammatory process is an important part of both arms of the immune system.
- Acute inflammation, characterized by influx of neutrophils into injured tissue, is one of the earliest events in an innate immune response.
- This process may be enhanced by components of the adaptive immune response, such as antibodies and cytokines.
- Inflammation is central to maintaining tissue homeostasis.
- While some degree of inflammatory responses limiting the progression of an insult is beneficial, exaggerated or prolonged inflammation or the lack of an adequate inflammatory response can lead to disease.
Cells of the Inflammatory Response: Inflammation Results in the Importation and Activation of Effector Cells at the Infection Site
- FIGURE 1 shows a schematic representation of the components and sequential involvement of cells that participate in the inflammatory response together with their known functions.
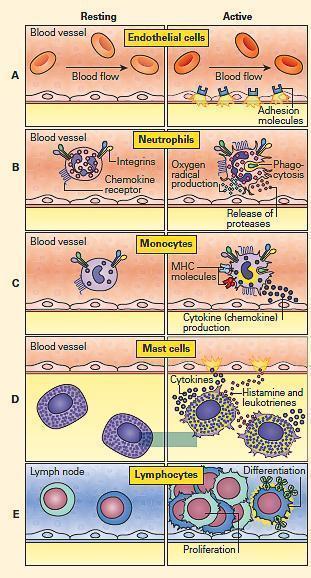
Figure 1: Schematic representation of cells involved in the inflammatory response and their functions. Panel A: Endothelial cells; Panel B: Neutrophils; Panel C: Monocytes; Panel D: Mast cells; and Panel E: Lymphocytes. (Adapted from a figure kindly provided by Peter Gentile). [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
ENDOTHELIAL CELLS
- Endothelial cells regulate leukocyte access to the tissues (FIGURE 1A).
- During an acute or chronic inflammatory response, leukocytes usually exit the circulation and enter surrounding tissue via post-capillary venules.
- The endothelial cells that line the lumen of these vessels are active participants in this process.
- The endothelial cell layer is one cell thick, and tight junctions exist between cells, that normally prevent the leakage of fluid into surrounding tissues.
- Following activation, endothelial cells undergo structural and functional changes that allow leakage of fluid and plasma proteins into the surrounding tissue (FIGURE 1).
- In addition, they upregulate cytokines and adhesion molecules, important for the tethering and tight adhesion of leukocytes.
NEUTROPHILS
- Neutrophils are the most abundant leukocytes and the first to arrive during an acute inflammatory response (FIGURE 1B).
- These cells are potent killers and contain multi-lobed nuclei, few organelles, and abundant granules.
- On activation, the cytoplasmic granules can be rapidly mobilized to the cell surface, simultaneously releasing their contents into the microenvironment and dramatically changing the array of receptors on the cell surface (FIGURE 1B).
- The granules contain large quantities of degradative enzymes and other antimicrobial products.
- Other granules can generate toxic oxygen radicals commonly referred to as reactive oxygen species (ROS).
- ROS not only destroy foreign invaders, but also may damage host tissue.
- Neutrophils also can engulf foreign microbes and cellular debris, i.e., phagocytosis, and this function can be enhanced by opsonins, i.e., C3b and antibody.
- Neutrophils are activated by cytokines and by immune complexes, which have the ability to enhance their killing mechanisms.
MONOCYTES/MACROPHAGES
- Monocyte/macrophages play a key role in chronic inflammatory responses (FIGURE 1C).
- Once a monocyte leaves the circulation and becomes activated, it differentiates into a highly phagocytic cell called a macrophage.
- Some examples of specialized macrophages include Kupffer’s cells in the liver, microglia in the CNS, and alveolar macrophages in the lungs.
- Like neutrophils, monocytes can phagocytose microbes and cellular debris, release degradative enzymes and produce toxic oxygen radicals, i.e., the ROS. 3
- However, macrophages are also highly anabolic cells that synthesize large quantities of many products such as cytokines and growth factors (FIGURE 1C).
- Macrophages also play an important role in the development of specific immunity during an adaptive immune response.
- Following the phagocytosis and breakdown of microbes, macrophages can function as antigen-presenting cells (APCs) and present the microbial fragments (in the context of MHC molecules) to T lymphocytes.
- Activated T lymphocytes can then help generate an antibody (i.e., Th2 cells interacting with B cells) or a cytotoxic T cell response.
- In either case, the response is specific for the microbe that was initially presented in the MHC of the macrophage.
- When foreign materials cannot be eliminated, macrophages together with lymphocytes participate in the formation of a granuloma.
MAST CELLS
- Mast cells are derived from bone marrow, but when mature, live within tissue, near nerves and blood vessels (FIGURE 1D).
- Together with basophils, they are referred to as mediator cells, and their strategic locations in tissue and blood allow them to respond rapidly to inciting stimuli by the release of preformed mediators, e.g., histamine and newly synthesized mediators, e.g., leukotriene metabolites.
- Mast cells are packed with granules that primarily contain histamine (an important preformed mediator that causes increased vascular permeability) and proteases.
- The inflammatory response is also enhanced through the activity of leukotrienes and cytokines such as TNF-α (FIGURE 1D).
- Mast cells and basophils express the high affinity IgE receptor—Fc epsilon receptor I (FcɛRI)—on their surface, and have been most extensively studied regarding their role in allergy.
- Mast cells also actively produce mediators that participate in the inflammatory response.
- Many inflammatory stimuli such as complement activation products, bacterial surfaces, and chemokines stimulate mast cell degranulation.
LYMPHOCYTES
- Lymphocytes also participate in chronic inflammation (FIGURE 1E).
- Although the proliferation and differentiation of lymphocytes, especially B cells, occurs primarily in lymphoid tissues rather than the infection site, lymphocytes play important roles in inflammation, particularly in chronic inflammation.
- Some subsets of lymphocytes carry out cytotoxicity (CD8+T cells and natural killer [NK] cells) while others are important for cytokine production, e.g., Th1 cells (interleukin-2 [IL-2], Interferon–gamma [IFN-γ], and tumor necrosis factor beta [TNF-β]) or Th17 cells (interleukin-17, –21, and –22 [IL-17, IL-21, and IL-22]).
- The primary function of cytotoxic cells is to seek out and destroy virus-infected cells and cancerous cells at the infection or tumor site.
- The production of pro-inflammatory cytokines by the Th1 and Th17 subpopulations results in the up-regulation of a variety of cellular events (e.g., chemotaxis and activation of macrophages), allowing them to kill phagocytosed pathogens more effectively.
- The production of IL-4, IL-5, IL-6, IL-9, and IL-13 by CD4+ Th2 cells leads to activation of B cells with subsequent antibody production or can result in recruitment of eosinophils (i.e., IL-5).
Molecules involved in Inflammation
-
- The innate immune response constitutes the first response to a foreign molecule or infectious agent
- After overcoming the constitutive and physiological barrier, the invader next encounters in plasma and body fluids a variety of soluble molecules, whose immediate activation can provide a protective defense
- These include molecules of the coagulation, fibronolytic and kinin cascades and from complement activation.
- The next defensive component of innate immunity occurs within 1-4 hours, via production of a family of molecules that activate the inflammatory response, involving epithelial, endothelial, dendritic and mast cells.
- This includes the up-regulation of adhesion molecules on endothelial cells of blood vessels and on phagocytic cells (neutrophils, monocytes, eosinophils).
- This leads to activation of these cells and other inflammatory cells such as mast cells and basophils, recruitement of NK cells and facilitates migration of some of these cells from blood into tissues.
- Usually the innate immune response is adequate to eliminate the foreign invader. Within 96 hours, the the adaptive immune response follows, with activation of antigen-specific effector cells and memory cellsthat can respond faster upon subsequent encounter with the same or closely related foreign substance.
- Figure 2 shows the interactive relationships between the innate and adaptive immune responses to a microbial infection and how innate and adaptive responses are totally inter-related.
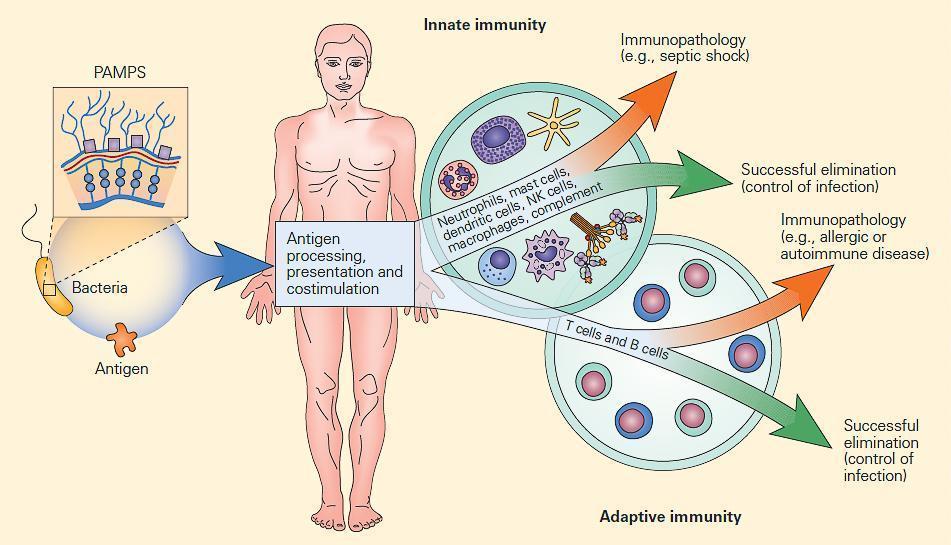
Figure 2: Schematic representation of the interactive relationships between the innate and adaptive immune responses to microbial infection. Infectious agents can be considered as mosaics of PAMPs and antigens. The innate immune response is induced early by PAMPs and assists the antigen-driven adaptive immune responses through antigen processing, presentation, and co-stimulation. Although both immune responses are usually effective in controlling infections, at times an uncontrolled or exaggerated response can result in self-damage from an uninhibited innate immune response (e.g., the systemic inflammatory response syndrome [SIRS]) or can lead to immunopathology from an unrestrained adaptive immune response (e.g., autoimmune disease). [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
The innate immune system recognizes PAMPs
-
- PAMPs are pathogen-associated molecular patterns, shared by large groups of microorganisms but absent from the host. They include proteins, polysaccharides (LPS) and glycolipids expressed on viruses and bacteria. They can also be released in soluble form or exist as intracellular components such as nucleic acids (viral single- or double-stranded RNA, CpG rich sequences).
- The receptors for PAMPs in the host are the PRR (pattern recognition receptors) which can be found as soluble molecules in body fluids (e.g. C-reactive protein (CRP) or mannose-binding protein, MBP) or as membrane-associated sensors at the cell surface or intracellularly.
- Activation of PRRs by PAMPs results in cell activation through production of molecules such as cytokines
- Interest in innate immunity started after the discovery of Toll-like receptors (TLRs) able to recognize different PAMPs.
Table 1: Summary of human Toll Like Receptors (TLRs)
| Receptor | Ligand(s) | Pathogen |
|---|---|---|
| TLR 1 | Triacyl lipoproteins | Gram-negative bacteria, mycobacteria |
| Lipoproteins | All bacteria | |
| Lipoteichoic acids | Gram-positive bacteria | |
| TLR 2 | Lipoarabinomannan | Mycobacteria |
| Zymosan | Fungi | |
| Glycosylphosphatidylinositol | Protozoans | |
| TLR 3 | Double-stranded RNA | Viruses |
| Lipopolysaccharides | Gram-negative bacteria | |
| TLR 4 | Lipopeptides | Gram-negative bacteria |
| Viral glycoproteins | Viruses | |
| TLR 5 | Flagellin | Bacteria |
| Diacyl lipoproteins | Mycobacteria | |
| TLR 6 | Zymosan | Fungi |
| TLR 7 | Single-stranded RNA | Viruses |
| TLR 8 | Single-stranded RNA | Viruses |
| TLR 9 | Unmethylated CpG DNA | Bacteria |
| TLR 10 | Unknown | Unknown |
| TLR 11 | Profilin | Protozoans |
- Some of the TLRs are surface-bound and some are intracellular
- FIGURE 3 shows the cellular location of these receptors in a generic cell
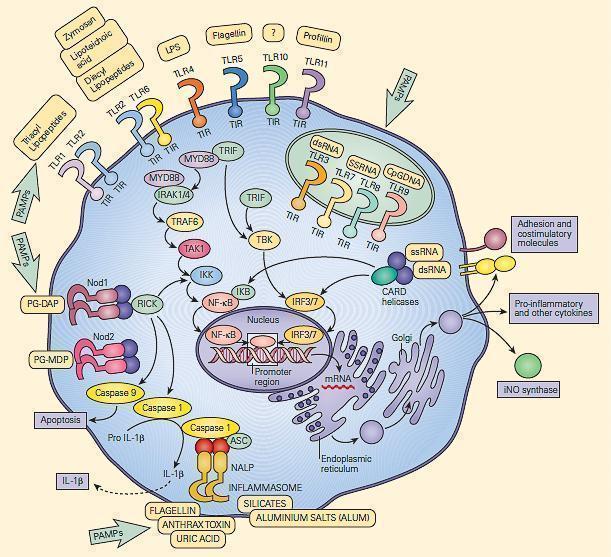
Figure 3: Schematic representation of the membrane-associated and intracytoplasmic location of the toll-like receptors (TLRs) and the critical role of the recognition and signaling pathways of the TLRs and the inflammasome. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
The difference between Innate and adaptive immune responses resulting in pathogen elimination
- The innate immune response is induced early by pathogen-associated molecular patterns (PAMPs)
which are recognized by the pattern recognition receptors (PRRs), including Toll-like Receptors (TLRs). - PRRs promote phagocytosis and production of inflammatory mediators which attract leukocytes to the infection site. Then, integrated events occur between inflammatory cells and the vascular endothelium via Selectins, Integrins and their Ig superfamily
- The antigen-driven adaptive immune response requires polymorphic MHC class I and Class II molecules to present antigen to TCR and generate a lasting and specific immune response.
Cellular and receptor:ligand events in inflammation
- Initial contact mediated by selectins (L-,E-, and P) and their glycosylated ligands leading to loose and transient adherence of inflammatory cells (rolling) to the vascular endothelium.
- Subsequently, Integrins and their Ig superfamily ligands mediate tighter adhesion, locomotion and transendothelial migration.
- Inflammation is characterized by the capture of leukocytes by loose bonding of the leukocyte to the endothelial cell
- Because the bond between selectins and their carbohydrate ligands is transient and of low affinity, the leukocytes are actually rolled over the vessel wall by the force of blood flow passing over
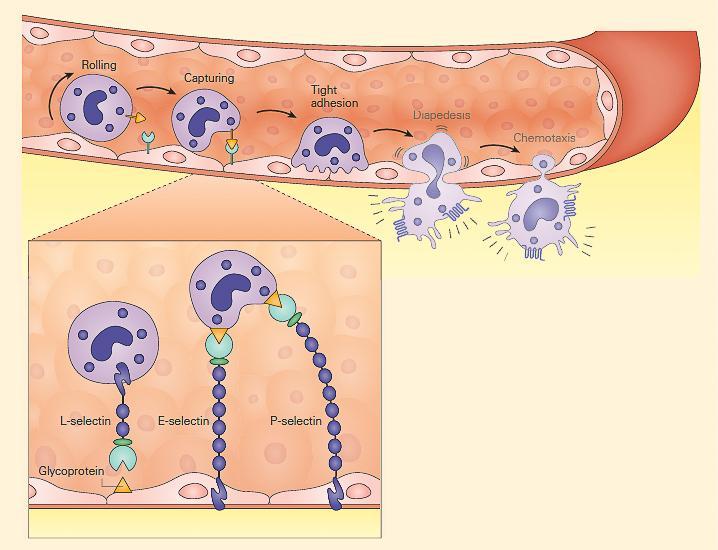
Figure 4: Schematic representation of the capture phase of the leukocyte-endothelial interaction showing the loose binding of selectins to glycoproteins. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- Another set of adhesion molecules that participate in the leukocyte-endothelial interaction involved in inflammation is the immunoglobulin superfamily of molecules
- The predominant molecules of this family that play a role in leukocyte endothelial adhesion are intercellular adhesion molecules 1 and 2 (ICAM-1, and ICAM-2), and vascular cell adhesion molecule 1 (VCAM-1) – shown in FIGURE 5
- Variable numbers of these molecules are expressed on the surface both constitutively and after cellular stimulation
- For these molecules to function properly and bind to integrin ligands, integrins first need to become activated; this occurs when the cell that bears the integrin is stimulated sufficiently by one of the proinflammatory mediators or chemoattractants described previously
- When activated, the integrin molecule can form a high affinity bond with its counter-ligand, the strength of which causes the leukocyte to stop rolling and adhere tightly.

Figure 5: Schematic representation of the tight adhesion phase of the leukocyte-endothelial cell interaction showing the firm binding brought about by the interaction of integrins on the leukocyte membrane with ICAMs on the endothelium. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Role of Selectins in Inflammation
- During inflammation, E-selectin plays an important part in recruiting leukocytes to the site of injury.
- The local release of cytokines IL-1 and TNF by damaged cells induces the overexpression of E-selectin on endothelial cells of nearby blood vessels.
- Leukocytes in the blood expressing the correct ligand will bind with low affinity to E-selectin, causing the leukocytes to “roll” along the internal surface of the blood vessel as temporary interactions are made and broken.
- As the inflammatory response progresses, chemokines released by injured tissue enter the blood vessels and activate the rolling leukocytes, which are now able to tightly bind to the endothelial surface and begin making their way into the tissue.
- P-selectin has a similar function, but is expressed on the endothelial cell surface within minutes as it is stored within the cell rather than produced on demand.
- L-selectin is expressed on neutrophils, a subset of T cells, and monocytes.
- Unlike either P-selectin and E-selectin, L-selectin is constitutively expressed.
- In addition, neutrophils shed L-selectin in response to chemotactic stimuli (including IL-8, C5a, and the surrogate bacterial chemoattractant N-formyl-methionyl-leucyl-phenylalanine), but not in response to immune complexes.
- L-selectin binds to CD34 and other glycosylated proteins on the surface of vascular endothelial cells.
| Adhesion molecule | Specific cellular location | Ligand(s) | Primary function |
|---|---|---|---|
| Selectins (transient interaction) | |||
| P-selectin | Platelet and endothelial cell surface expression after stimulation | Glycoproteins | Mediate cellular margination and rolling |
| E-selectin | Exclusively expressed on stimulated endothelial cells | Glycoproteins | |
| L-selectin | Constitutively expressed on neutrophils, monocytes, and a T cell subset | Glycoproteins | |
| Shed from neutrophils after stimulation with chemoattractants | |||
| Immunoglobulin superfamily (strong interaction) | |||
| ICAM-1 (CD54) | Low-level constitutive endothelial cell expression | LFA-1, Mac-1 | Play a major role in leukocyte-endothelial adhesion |
| Increased endothelial cell expression with stimulation | |||
| ICAM-2 (CD102) | High-level constitutive endothelial cell expression | LFA-1, Mac-1 | |
| No increase with stimulation | |||
| ICAM-3 (CD50) | Lymphocytes | LFA-1 | |
| ICAM-4 (CD242) | Erythrocytes | LFA-1, Mac-1 | |
| VCAM-1 (CD106) | No constitutive endothelial cell expression | VLA-4 | |
| Increased endothelial cell expression with stimulation | |||
| Integrins (strong interaction) | |||
| LFA-1 (CD11a/CD18b) | Lymphocytes | ICAM-1, ICAM-2, ICAM-3, ICAM-4 | Leukocyte migration, phagocytosis, and growth and development |
| Mac-1 (CR3, CD11b/CD18b) | Neutrophils and monocytes | ICAM-1, ICAM-2 | |
| gp 150/95 (CD11c/CD18b) | Neutrophils and monocytes | ICAM-1, ICAM-2 | |
| VLA-4 (very late antigen-4) | Lymphocytes, monocytes, and eosinophils | VCAM-1 |
Diapedesis and Chemotaxis of Leukocytes Toward an Inflammatory Stimulus
- Following tight adhesion, leukocytes migrate for a short distance along the endothelial surface until they arrive at a junctions between endothelial cells.
- The leukocytes then squeeze between endothelial cells to gain access to surrounding tissue.
- This process, called diapedesis, is thought to be mediated by adhesion molecules named platelet/endothelial cell adhesion molecule-1 (PECAM-1) and CD99.
- This movement of cells is part of the overall process of chemotaxis in which the movement of cells is directed by chemoattractants such as products of bacterial invasion or the complement components C3a and C5a.
Inflammation Is Normally Controlled by Negative Feedback Mechanisms
- Inflammation normally leads to recovery and healing.
- However, if targeted destruction and assisted repair are not properly phased, acute inflammation can lead to persistent chronic tissue damage mediated by leukocytes, lymphocytes, or collagen.
- Inflammation may be considered in terms of its checkpoints, where a cascading set of signals drives each to escalate.
- However, these “go” signals trigger “stop” signals, and, paradoxically, molecules responsible for mediating the inflammatory response can also suppress it, depending on timing and context.
- Some examples of these inhibitory mechanisms include delayed macrophage production of an IL–1 receptor antagonist (IL-1ra), TNF induction of TNF receptor (TNFR), shedding and competition of the released receptors with cell surface receptors for TNF, production of anti-inflammatory cytokines IL-10 and TGF-β, and CNS suppression of inflammation.
- The non-inflammatory state does not arise passively from an absence of inflammatory stimuli; rather, maintenance of homeostatic balance in health requires the positive actions of specific gene products to suppress reactions to potentially inflammatory stimuli that do not warrant a full response.
Cell Death and Inflammation: Apoptosis, Autophagy, Oncosis and Pyroptosis
- One of the outcomes of inflammation is cell death.
- Cells can die through distinct biochemical pathways that produce different morphological and physiological outcomes.
- There are four basic terms that have been used to describe these various outcomes: apoptosis, autophagy, oncosis and pyroptosis.
- Apoptosis and autophagy are mechanisms that maintain normal intracellular homeostasis and do not lead to inflammation
- This is in contrast to oncosis and pyroptosis, which are associated with release of intracellular contents, necrosis and inflammation.
- The stimulation of inflammation by pyroptosis is mediated by activation of the inflammasome.
Apoptosis
- Apoptosis (from the Greek, denotes a “falling off,” as leaves from a tree) is perhaps the most widely recognized term to describe programmed cell death without inflammation.
- In this scenario, cells die as a result of the requirement for particular cysteine dependent aspartate-specific proteases (caspases) which produce an orchestrated disassembly of the cell
- The contents of apoptotic cells are packaged into membrane-enclosed structures called apoptotic bodies, which are recognized and taken up by macrophages, i.e., phagocytosis, and removed in vivo, resulting in an absence of inflammation
- Apoptosis is the physiological mechanism by which most normal cells are regularly removed and eliminated
Autophagy
- Autophagy, or autophagocytosis (from the Greek, to denote self-eating) is a second non-necrosis inducing pathway in which the cell literally degrades its own components through the lysosomal machinery
- The most well-known mechanism of autophagy involves the formation of membrane-surrounded structures enclosing targeted regions of the cell called autophagic vacuoles, which separate their contents from the rest of the cytoplasm.
- The resultant vesicle then fuses with a lysosome and subsequently degrades its contents.
- Like apoptosis, autophagy represents a tightly-regulated process that plays a normal part in cell growth, development, and homeostasis, helping to maintain a balance between the synthesis, degradation, and subsequent recycling of cellular products.
- Autophagy has been identified as a route by which intracellular infectious agents or tumor-derived and self antigens are transferred from the cytoplasm to the lysosome where they are degraded and delivered to MHC-II molecules for presentation to CD4+ T cells.
Oncosis
- The term oncosis is a type of cell death in which the cell undergoes swelling (from the Greek, onkos, which means swelling) as a counterpoint to apoptosis.
- Oncosis is defined as a prelethal pathway leading to cell death accompanied by cellular swelling, organelle swelling, blebbing, and increased membrane permeability
- The process of oncosis ultimately leads to depletion of cellular energy stores and failure of the ionic pumps in the plasma membrane.
- Oncosis may result from toxic agents that interfere with ATP generation or processes that cause uncontrolled cellular energy consumption.
Pyroptosis
- Pyroptosis (from the Greek pyro, which relates to fire or fever, and ptosis, a “falling off”) is the most recently described mechanism of cell death associated with features of cell lysis and release of inflammatory cellular contents
- It is a pathway morphologically and mechanistically distinct from other forms of cell death in which caspase 1 dependence is a defining feature.
- Unlike apoptosis, which involves caspase 3, caspase 6 and caspase 8, in pyroptosis caspase 1 is the enzyme that mediates this process of cell death.
- Pyroptosis features rapid plasma-membrane rupture and release of proinflammatory intracellular contents.
- Pyroptosis is not only involved in host protection against infection but can also induces pathological inflammation.
Soluble Mediators Regulate the Inflammatory Response
- In addition to cell-associated mediators, there are several substances that circulate in the blood as soluble mediators of inflammation. Shown in Table 3 is a list of soluble mediators that regulate the inflammatory response together with their specific functions.
| Mediator | Examples | Functions |
|---|---|---|
| Cytokines | IL-1, IL-6, TNF-α | Vasodilatation, increased endothelial cell adhesion molecule expression |
| Fever, acute phase protein release from liver, neutrophil release from bone marrow | ||
| IFN-γ | More efficient macrophage killing of phagocytosed pathogens | |
| Increased MHC expression | ||
| IL-10, TGF-β | Anti-inflammatory effects | |
| Immune complexes | Antigen bound to IgM or IgG | Complement activation |
| Macrophage and neutrophil production and release of toxic metabolites | ||
| Increased phagocytosis | ||
| Complement anaphylatoxins | C5a | Mast cell degranulation, chemotaxis, and activation of leukocytes |
| Proteases | Serine proteases | Catabolize microbial proteins |
| Metalloproteases | Digest host proteins | |
| Thiolproteases | ||
| Acid proteases | ||
| Chemokines | IL-8, macrophage chemotactic protein (MCP) | Leukocyte chemotaxis |
| Arachadonic acid metabolites | Thromboxane | Platelet aggregation |
| Prostaglandins | Vasodilation | |
| Leukotrienes | Pain stimulation | |
| Leukocyte chemotaxis |
Some cytokines, such as tumor necrosis factor alpha (TNF-α), IFN-γ, interleukin-6 (IL-6), and interleukin-1 (IL-1), are thought to have predominantly pro-inflammatory effects, while other cytokines, such as interleukin-4 (IL-4), interleukin-10 (IL-10), and transforming growth factor-beta (TGF–β), are thought to have predominantly anti-inflammatory effects. Currently, a paradigm exists in rheumatology stating that an imbalance between the relative amounts of “pro-inflammatory” and “anti-inflammatory” cytokines can trigger the development of inflammatory/autoimmune diseases. This paradigm, however, may be oversimplified as it has become clear that whether or not any particular cytokine exhibits pro- or anti-inflammatory properties depends largely upon the timing and context of its appearance during the immune response.
- Cytokines are a large group of proteins that regulate the activation, proliferation, and differentiation of immune cells.
- Although leukocytes are the major sources of cytokines, many other cell types can also synthesize cytokines as well.
- FIGURE 6 illustrates some of the leukocyte-derived cytokines involved in inflammatory arthritis.
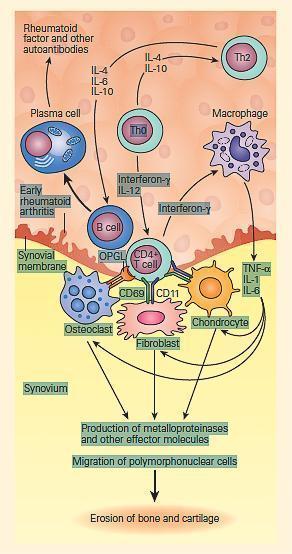
FIGURE 6: Schematic representation of cytokine signaling pathways involved in inflammatory arthritis. Antigen-activated CD4+ T cells stimulate macrophages to produce interleukin-1 (IL-1), interleukin-6 (IL-6), and TNF-α and synovial fibroblasts to secrete matrix metalloproteinases and other effector molecules. This occurs through signaling pathways mediated by the cell surface receptors CD69 and CD11 as well as through the release of soluble mediators such as interferon-γ. IL-1, IL-6, and TNF-α are the principal cytokines that drive inflammation in rheumatoid arthritis. Activated CD4+ T cells also stimulate B cells through cell surface contact with the cytokines IL-4, IL-6, and IL-10 released from Th2 helper lymphocytes. Although rheumatoid factor and other autoantibodies are known to be produced by plasma cells, their precise pathogenic role in RA is unknown but may participate in inflammation through the activation of complement and formation of immune complexes. Activated CD4+ T cells also express osteoprotegerin ligands (OPGL) that stimulate osteoclastogenesis that may participate in the joint damage of rheumatoid arthritis. (Adapted from Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 200;344: 907–16.) [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Inflammasomes, the Innate Immune System, and IL-1
- The innate immune system provides a critical protective function during the initial phase of microbial invasion and a homeostatic safeguard for the removal of worn out cellular components during the healthy physiologic state.
- As mentioned above, it does so using germline-encoded PRRs that not only detect danger signals generated by microbes but also by endogenous products of injured tissues that also can initiate inflammatory responses.
- A major pathway utilized by the innate immune system to carry out these functions relies on the production of potent pro-inflammatory cytokines, most notably IL-1β, through specialized cytoplasmic multimeric protein structures called inflammasomes, found within phagocytic cells of the innate immune system, particularly dendritic cells and macrophages.
- The inflammasome (from the Latin word inflammare, which means to set on fire; from the Greek word soma, which means body) is a danger-sensing complex that triggers innate immunity-linked inflammatory processes by induction of pyroptosis.
- In the detection of external and internal pathogenic signals, the innate immune system utilizes five types of PRRs: (1) TLRs, (2) C-type lectin receptors (CLRs), (3) nucleotide-binding oligomerization domanis (NOD)-like receptors (NLRs), (4) RIG-like helicases (RLHs), and (5) DNA sensors absent in melanoma 2 (AIM2) and (DNA-dependent activator of IFN (DAI)-regulatory factors) (Table 4).
| Pattern-recognition receptors (PRRs) | Danger-inducing ligands (PAMPs and DAMPs) | Function |
|---|---|---|
| Membrane-associated PRRs | ||
| Toll-like receptors (TLRs) | Molecular moieties found on bacteria and viruses and from damaged and necrotic cells | Initiate signaling pathways, e.g., NF-κB, to modulate gene expression; indirectly activates the inflammasome |
| C-type lectin receptors (CLRs) | Carbohydrate moieties on microbes or cell surfaces | Serve as antigen receptors on APCs but also regulate the migration of dendritic cells and their interaction with lymphocytes |
| Nonmembrane-associated PRRs | ||
| NOD-like receptors (NLRs) | Intracellular bacterial or viral components | Discriminate between pathogenic and commensal bacteria; directly activates the inflammasome |
| RIG-like helicases (RLHs) | Viral RNA | Promote type 1 interferon synthesis in cells infected by viruses to control the infection |
| DNA sensors (AIM2 and DAI) | Double-stranded DNA | Direct binding to double-stranded cytosolic DNA and activate inflammasome |
- These receptors recognize conserved molecular moieties found on a wide variety of microbes (“non-self”) as well as those present in disorganized or modified self-structural proteins (“modified self”).
- Collectively, these danger-detecting PRRs recognize innate-stimulating triggers known as PAMPs or danger-associated molecular patterns (DAMPs) or alarmins, respectively.
- The basic structure of the inflammasome consists of three major components: (1) an NLR, (2) an adapter protein, and (3) a caspase.
- The most extensively studied inflammasome is the NLRP3 inflammasome, and most of the currently recognized monogenic clinical disorders involve disruption of this pathway. Shown in FIGURE 5 is a schematic representation of the tripartite structure of the NLRP3 inflammasome.
Comparison between autoinflammatory and autoimmune diseases
- The term autoinflammation is now used interchangeably with innate immune-mediated inflammation, and it is possible to draw a comparison between the autoinflammatory diseases and the autoimmune diseases (Table 5).
| Distinguishing feature | Autoimmune diseases | Autoinflammatory diseases |
|---|---|---|
| Arm of immunity affected | Adaptive immunity | Innate immunity |
| Genetic basis | Monogenic and polygenic disorders of adaptive immune function | Monogenic and polygenic disorders of innate immune function |
| Specific dysregulated component | Primary dysregulation of classical MHC-based, antigen-dependent T cell responses | Primary dysregulation of innate immune system processing and secretion of pro-inflammatory cytokines, IL-1β, IL-18, and others |
| Resultant secondary contribution of inflammatory responses | Resultant primary contribution of inflammatory responses | |
| Effector mechanisms involved | Injury mediated by activation of CD4 subpopulations (Th1, Th2, Th17, and Treg) together with other innate effector cells (macrophages, mediator cells, NK cells via cytokine production) | The pathological abnormality in autoinflammatory diseases is a failure to control processing and secretion of IL-1β and other pro-inflammatory cytokines in patients with these diseases |
| Tissue destruction, mediated directly by cytotoxic CD8 T cells | ||
| T cell-dependent B cell autoantibody production | ||
| Examples of diseases | Organ-specific autoimmune diseases | Familial Mediterranean fever |
| (Celiac disease, Graves' disease, type 1 diabetes, Addison's disease, autoimmune thyroiditis) | Neonatal-onset multiple system inflammatory disease | |
| Systemic autoimmune diseases | Systemic-onset juvenile idiopathic arthritis (JIA) | |
| (SLE, RA) | ||
| Predominant symptoms | Fever | Fever |
| Maculopapular rash | Urticarial rash | |
| Joint involvement (arthritis or arthralgias) | Pyogenic arthritis | |
| Specific organ involvement | Pyoderma gangrenosum | |
| Neurologic involvement |
- The autoimmune diseases refer to disorders characterized primarily by aberrant adaptive immune responses, in contrast to the autoinflammatory diseases, which are primarily disorders of the innate immune system.
- The majority of both conditions are transmitted as monogenic and polygenic disorders of either the adaptive or innate immune systems, respectively.
- Examples of the autoimmune diseases include both the organ-specific and systemic autoimmune diseases, characterized both by aberrations of classical MHC-based antigen-dependent T cell interactions, injury mediated by activation of CD4 subpopulations (Th1, Th2, Th17, and Treg), together with other innate effector cells (macrophages, mediator cells, and NK cells via cytokine production). Tissue destruction in the autoimmune diseases is primarily mediated directly either by cytotoxic CD8 T cells or by T cell-dependent B cell autoantibody production.
- The autoinflammatory diseases, on the other hand, represent a group of monogenic and polygenic diseases that can present clinically with recurrent inflammation and unexplained fevers as part of their phenotype (Table 5).
- Examples of the innate-mediated autoinflammatory diseases include familial Mediterranean fever, neonatal-onset multisystem inflammatory disease, and systemic-onset juvenile idiopathic arthritis (JIA). Patients with these diseases suffer from chronic fever, urticarial rashes, arthritis, and neurologic involvement. Rapid reversal of disease severity generally occurs after the blocking of IL-1β activity by immune modulating agents.
- The pathological abnormality in many of the autoinflammatory diseases appears to be a failure to control processing and secretion of IL-1β and other pro-inflammatory cytokines. The processing and secretion of IL-1β is controlled by caspase-1, an intracellular protease that cleaves the IL-1β precursor, and pro-IL-18, into active cytokines and is a component of the inflammasome.
- Recent studies have shown that activation of the IL-1β pathway, which is a common mechanism in the pathogenesis of autoinflammatory diseases, is a unifying factor in these diseases. Constitutive increases in the secretion of IL-1β and IL-18 have been shown in macrophages from NOMID/CINCA and MWS patients, suggesting that mutations in NLRP3 increase production of these pro-inflammatory cytokines.
Autoinflammatory Diseases
- Many rare monogenic and polygenic diseases are thought to be involved in defects of the pathway synthesis of IL-1β, resulting in its overproduction and the consequent pathogenic chronic or periodic inflammatory attacks. For this reason, they have been called autoinflammatory diseases.
- A new molecular/functional classification of autoinflammatory diseases has been proposed and is shown in Table 6.
- In this functional classification, the first type correspond to defects found in the NLRP3 inflammasome and have been termed cryoparinopathies or cryopyrin-associated periodic syndromes (CAPS).
- CAPS result in periodic episodes of inflammation due to the overproduction of IL-1β after innocuous stimulation from cold, i.e., familial cold autoimmune syndrome (FCAS) or others, for reasons still not understood completely, i.e., neonatal onset multisystem inflammatory disease (NOMID) (see Bellanti, Chapter 19 Annex).
| Disease | Example of disease | Gene/(chromosome)/product |
|---|---|---|
| Type 1: IL-1 β activation disorders (inflammasomopathies) | ||
| Intrinsic | Familial cold autoimmune syndrome (FCAS) NOMID/CINCA | NLRP3/CIAS1 (1q44) |
| Muckle Wells | ||
| Extrinsic | FMF | MEFV (16p13.3)/pyrin (marenostrin) |
| PAPA | PSTPIP1 (15q24-25.1) | |
| DIRA | IL1RN/IL1Ra | |
| CRMO/SAPHO, | Complex | |
| HIDS | MVK (12q24)/mevalonate kinase | |
| Acquired or complex | Gout | Complex/uric acid |
| Type 2 diabetes mellitus | Complex/hyperglycemia | |
| Fibrosing disorders (silicosis, asbestosis) | Complex/asbestos and silica | |
| Type 2: NF- κ B activation disorders | Crohn's disease | NOD2 (16p12)/NOD2(CARD15) |
| Blau syndrome | NOD2 (16p12)/NOD2(CARD15) | |
| Familial cold autoimmune syndrome (FCAS2) | NLRP12 (19q13.4)/NRLP12(NALP12) | |
| Type 3: Protein-folding disorders of the innate immune system | TNF receptor-associated periodic | TNFRSF1A (12p13)/TNFR1 |
| syndrome (TRAPS) | Complex | |
| Spondyloarthropathies | HLA-B (6p21.3)/HLA-B27 | |
| ERAP1 (5q15)/ERAP1 | ||
| Type 4: Complement disorders | Acquired hemolytic uremic syndrome (aHUS) | CFH (1q32)/Factor H |
| MCP (1q32)/MCP (CD46) | ||
| CFI (4q25)/Factor I | ||
| CFB (6p21.3)/Factor B | ||
| Age-related macular degeneration | CFH (1q32)/Factor H | |
| Type 5: Cytokine-signaling disorders | Cherubism | SH3-binding protein 2/SH3-binding protein 2 |
| Type 6: Macrophage activation | Familial hemophagocytic lymphohistitiocytosis (HLH) | UNC13D (17q21.1)/Munc13-4 |
| PRF1 (10q22)/Perforin 1 | ||
| STX11 (6q24.2)/Syntaxin 11 | ||
| Complex/virus | ||
| Chediak-Higashi syndrome | LYST (1q42.3)/ LYST (CHS1) | |
| Griscelli syndrome | RAB27A (15q21.3)/ RAB27A | |
| X-linked lymphoproliferative syndrome | SH2D1A (Xq25)/ SAP | |
| Hermansky-Pudlak syndrome | HPS1-8/ HPS1-8 | |
| Secondary HLH | Complex | |
| Atherosclerosis | Complex/cholesterol |
- Although the autoimmune diseases and the autoinflammatory diseases represent distinct polar entities, there are a number of intermediate entities with features of both, including psoriasis, ankylosing spondylitis, reactive arthritis, and Behcet’s disease.
- The driving force of this classification had its origins in the study of a group of relatively rare monogenic disorders but has led to a better understanding of more commonly encountered disorders that were diagnostic and therapeutic orphans, e.g., sarcoidosis, Crohn’s disease, and juvenile idiopathic arthritis.
- Table 7 shows a representation of the continuum of disorders comprising the autoimmune diseases and the autoinflammatory disorders.
| Intermediate diseases | Monogenic autoimmune diseases | |
|---|---|---|
| Monogenic innate immune diseases | ||
| CAPS | Psoriasis | ALPS |
| FMF | Ankylosing spondylitis | IPEX |
| HIDS | Reactive arthritis | APS-1 |
| TRAPS | Behcet's disease | |
| PAPA | ||
| Blau syndrome | ||
| Polygenic innate immune diseases | Polygenic autoimmune diseases | |
| Crohn's disease | Celiac disease | |
| Ulcerative colitis | Graves' disease | |
| Psoriatic arthritis | Type 1 diabetes | |
| Juvenile idiopathic arthritis | Addison's disease | |
| Environmentally induced autoinflammatory diseases | Autoimmune thyroiditis | |
| Gout | SLE | |
| Asbestosis | Rheumatoid arthritis | |
| Silicosis | ||
| Sarcoidosis (?) |
Key Points
- The autoinflammatory diseases refer to a group of disorders originally identified in a cluster of monogenic chronic or periodic inflammatory syndromes but that now have been broadened to include an increasing number of related polygenic and complex disorders that have as their common pathway inflammation initiated through the innate immune system, where supranormal synthesis of pro-inflammatory mediators, such as IL-1β, generates inflammation-induced tissue injury after exposure to a variety of both exogenous and endogenous stimuli.
- These stimuli range from a wide variety of external activators, which include cold, asbestos, silica, and alum adjuvants, to endogenous substances such as uric acid, glucose, and other endogenous metabolic products.
- A clear distinction should be made between the autoinflammatory diseases, which reflect defects of the innate immune system, and the autoimmune diseases, which express defects in the adaptive immune system.
- Although inflammation is the hallmark manifestation that distinguishes autoinflammatory diseases from autoimmune disorders, the two entities represent a continuing spectrum of disorders with considerable overlap between them.
- The aetiology of the autoinflammatory disorders has been biologically linked with several gene mutations involving key signaling pathways of the innate immune system that has provided the basis for a functional classification into six groups: IL-1β activation disorders (i.e., inflammasomopathies), NF-κB activation disorders, protein-folding disorders, complement disorders, signaling disorders in cytokine pathways, and macrophage disorders.
- A knowledge of the molecular mechanisms involved in the pathogenesis of the autoinflammatory disorders forms the basis for their current management with biologic modifying agents, e.g., IL-1β antagonists and TNF-α inhibitors, as well other emerging therapeutic modalities.





