





- Protracted or chronic bacterial infections are often caused by organisms that have an intracellular phase; this requires that they are adept at the penetration, evasion, and exploitation of the host immune response.
- The classic example of a chronic bacterial infection is seen with Mycobacterium tuberculosis, the causative organism of tuberculosis.
- These bacteria invade the host and set up a long-standing relationship by killing some cells and chronically infecting others, eliciting an unstable détente with host immunity that is typically detected only indirectly by demonstration of an antigen specific tuberculin skin test (TST) in response to antigens derived from the M tuberculosis, known as purified protein derivative.
- Figure 1 is a schematic representation of three outcomes of the interaction of a mycobacterium and a macrophage, illustrating either killing of the organism by the macrophage (Figure 1A), killing of the macrophage by the organism (Figure 1B), or survival of both the organism and the cell as seen in chronic infection (Figure 1C).
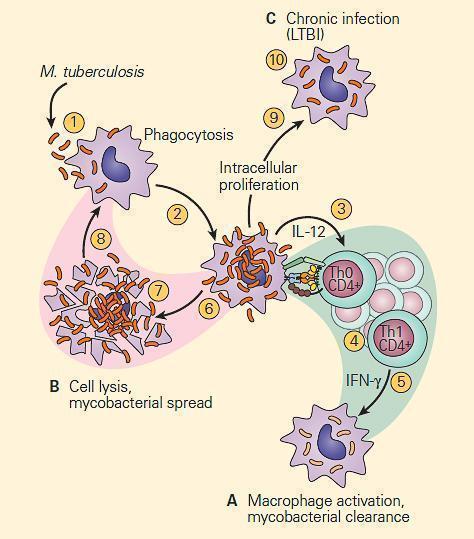
Figure 1. Schematic representation of three modes of interaction of the tubercle bacillus with a macrophage. Panel A: The macrophage kills the organism. Panel B: The organism kills the macrophage. Panel C: The organism takes up residence in the macrophage and establishes chronic infection, where both the tubercle bacillus and macrophage can survive together in a tenuous chronic infection. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- This third interaction between the host and the tubercle bacillus is thought to be the basis for latent tuberculosis infection (LTBI) which establishes an armistice between the host and the bacterium which can be broken as seen by reactivation of the active infection at a later point in life when the balance is tipped in favor of the bacterium
Latent tuberculosis (Latent TB) or Latent Tuberculosis Infection (LTBI)
- Latent tuberculosis is a clinical condition in which a patient is chronically infected with Mycobacterium tuberculosis, but does not show manifestations of active disease.
- It is believed (but not proven in the human) that the condition develops from an interaction between the tubercle bacillus and a macrophage shown in panel C of Figure 1
- Here, a tenuous armistice is established between the host and parasite which can be broken in later life in the form of reactivation of the tuberculosis infection.
- This can occur by any of a number of clinical conditions which suppress the host immune response, e.g. intercurrent viral infection, malnutrition, immunosuppressive therapy, aging or the use of anti-TNF-α agents.
- Patients with latent tuberculosis are not thought to be infectious, and it is not likely to contract TB from someone with latent tuberculosis.
- Since the main risk is that approximately 10% of these patients will go on to develop active tuberculosis at a later stage of their life, the identification and treatment of people with latent TB is an important part of controlling this disease.
- The recent development of highly specific assays which measure the release of interferon-γ from blood lymphocytes following in vitro stimulation with specific peptides of the M tuberculosis, i.e., the interferon gamma release assays (IGRAS) have not only been useful in identifying patients with LTBI but also in more accurately differentiating infections caused by M tuberculosis from those caused by other mycobacteria or following BCG vaccination which can be misdiagnosed by the less specific TST.
- Figure 2 are two types of mechanisms of bacterial clearance by phagocytes that are related to the virulence of an invading intracellular chronic disease producing bacterium such as the tubercle bacillus.
- The bacilli are not readily phagocytosed by opsonin-dependent uptake by neutrophils, but because of their intracellular location within macrophages, T cell-mediated immune mechanisms that enhance phagocytosis and intracellular killing by macrophages are of crucial importance.
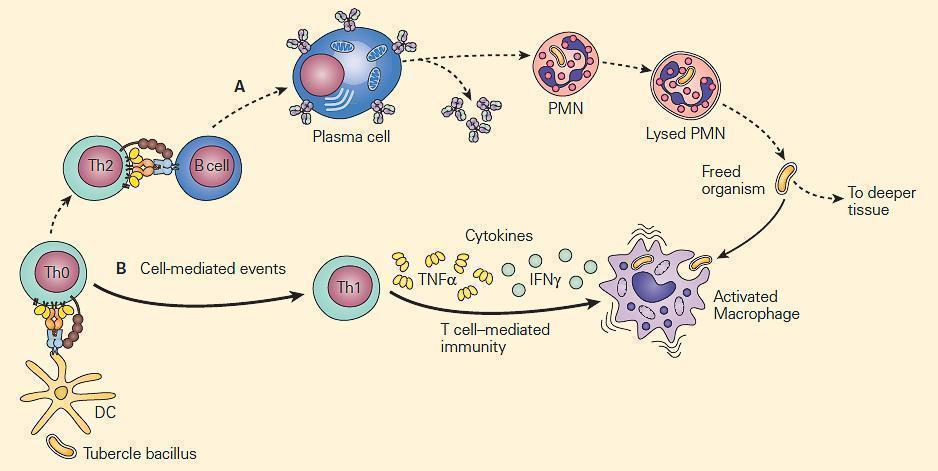
Figure 2. Schematic representation of the relative roles of antibody and cell-mediated events in the enhancement of phagocytosis during chronic bacterial infections. Panel A: PMNs show limited microbicidal activity. Panel B: The major cellular response in chronic infection is carried out through cytokine-induced stimulation of macrophages by TNF-α and IFN-γ. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
The T cell-Macrophage Interaction
- Two major T cell-macrophage axes have been described:
- 1) the IL-12/IFN-γ;
- 2) the IL-23/IL-17 pathways.
- Shown in Figure 3 is a schematic representation of the IL-12/IFN-γ axis.
- As can be seen there are many T cell-macrophage interactive cytokine pathways involved in chronic bacterial microbicidal activity.
- Deficiencies in any of these cytokine pathways have been described which contribute to increased mycobacterial disease susceptibility.
- Disruptions of the IL-12/IFN-γ pathway such as mutations of the interferon-γ (IFNγ) receptor, IL-12 receptor b1 (IL-12Rb1), and IL-12 p40 genes have been recognized.
- These mutations are associated with heightened susceptibility to disease caused by intracellular pathogens including nontuberculous mycobacteria, vaccine-associated Bacille Calmette Guerin (BCG) and Salmonella species.
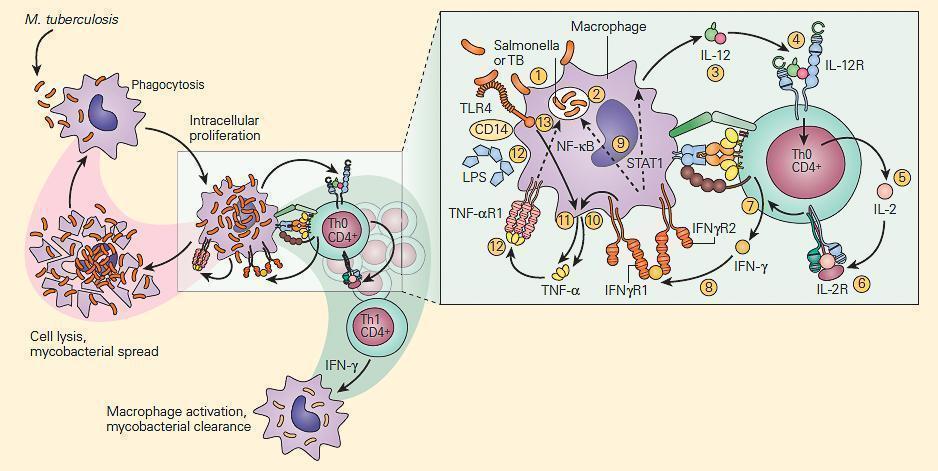
Figure 3. Schematic representation of the IL-12/IFN-γ pathway illustrating various activation pathways associated with the killing of intracellular bacterial organisms such as M. tuberculosis (TB) and Salmonella. Following the uptake of the TB by the macrophage (1) into a phagosome (2), the macrophage is stimulated to produce IL-12 (3). The binding of IL-12 to its IL-12R on a Th CD4+ cell (4) induces the synthesis of IL-2 (5), and after autocrine binding of IL-2 to its IL-2R (6) leads to the synthesis of IFN-γ (7). The binding of IFN-γ to its receptor (8) activates both the STAT1 and NF-kB pathways (9) to kill the TB and to further enhance IL-12 production; the IFN-γ-activated macrophage (10) and TLR4 activation by the TB (11) induce TNF-α production that, after binding to its receptor (12), leads to further enhancement of intracellular killing of the TB (13). [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- Shown in Figure 4 is a photograph of an infant with complete IFN-γR1 deficiency vaccinated with BCG who developed dissemination of the BGC (i.e., BCGosis) as a result of this deficiency.
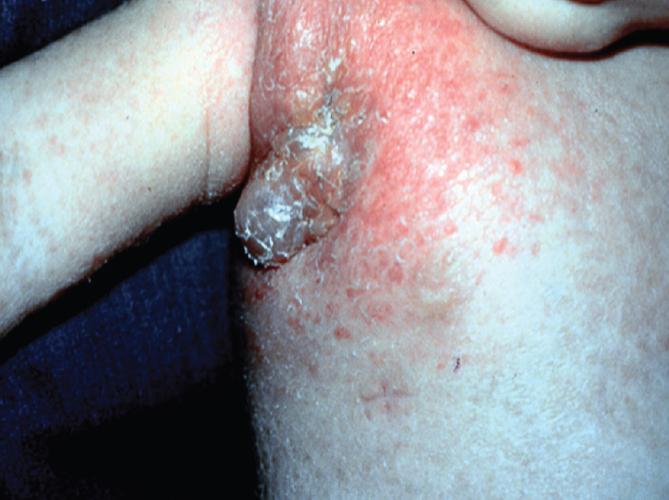
Figure 4. Photograph of an infant with complete IFN-gR1 deficiency who was vaccinated with BCG and who developed dissemination of the BGC (BCGosis) as a result of a defect of the IL-12/IFN-g pathway. The deficiency permitted the spread of the BCG infection to a regional lymph node with suppurative inflammation swelling and erythema in the right axilla. (Courtesy of Dr. Sergio Rosenzweig.) [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- The second T cell-macrophage axis important for maintenance of normal bacterial clearance is the IL-23/IL-17 pathway shown in Figure 5
- This pathway is particularly important at mucosal surfaces.
- IL-23 exerts its effects by activation of Th17 cells.
- The subsequent production of cytokines by these cells has opposing beneficial and detrimental effects; the production of IL-22 and IL-17 leads to the generation of antimicrobial peptides by epithelial cells while IL-17 can also increase granulocyte production and influx, promoting inflammation.
- The IL-23/17 axis appears to not only play an important protective role in antibacterial and antifungal immunity but also is an important regulator of immune responsiveness in various inflammatory diseases, e.g., Crohn’s disease, arthritis and allergic airway inflammatory disease.
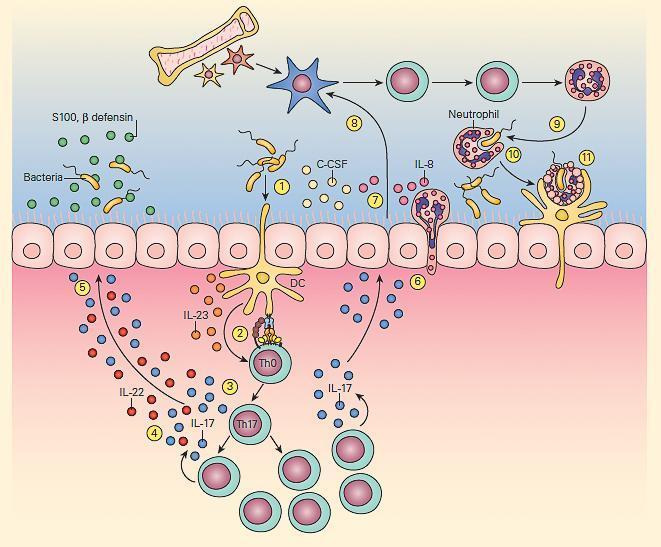
Figure 5. Schematic representation of the IL-23/17 axis involved in host antibacterial defense at a mucosal surface. Mucosal dendritic cells activated by bacterial pathogen (1) produce IL-23 (2), which then activates the production of resident memory Th17 cells from naive Th0 cells (3) to produce IL-17 and IL-22 (4), which then induce epithelial cells to secrete antimicrobial peptides, e.g., β-defensins and S-100 proteins (5). IL-17 also induces epithelial cells (6) to produce granulopoietic and chemotactic factors, e.g., G-CSF, IL-8 (7), which then leads to bone marrow granulocyte production (8). The granulocytes then home to the mucosal surface (9), where they phagocytose bacteria (10) and following their apoptotic cell death, their resultant apoptotic fragments are phagocytosed by dendritic cells (11). [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Granuloma formation
- The T cell-macrophage balance is further maintained by the creation of a structure referred to as a granuloma.
- The term “granuloma” is derived from the anatomical “granular” appearance of the lesions on gross examination and is a structure consisting mainly of epithelioid macrophages and other inflammatory and immune cells.
- The granuloma represents an abortive attempt of the host to wall off a foreign substance, which can neither be killed nor completely eliminated.
- The formation of a granuloma is highly dependent on proinflammatory cytokines e.g., TNF-α and IFN-γ (Figure 6).
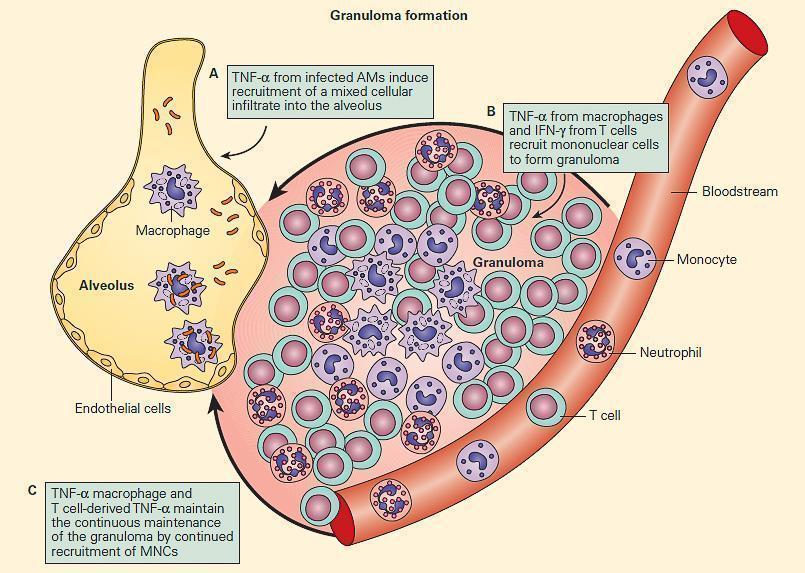
Figure 6. TNF acts at multiple steps in protective granuloma formation in response to M. tuberculosis infection. Panel A: TNF-α derived from infected alveolar macrophages initially induces recruitment of a mixed cellular alveolar and interstitial infiltrate. Panel B: Under the influence of TNF-α and T cell-derived IFN-γ, mononuclear cells accumulate to form a highly structured granuloma. Here, TNF-α and IFN-γ activate mycobactericidal pathways within macrophages and regulate excessive inflammation by inducing apoptosis of T cells. Panel C: Macrophage and T cell-derived TNF-α is necessary to continuously orchestrate the recruitment of mononuclear cells into granulomas in order to maintain effective containment of mycobacterial foci of infection. (Adapted from Ehlers S. Why does tumor necrosis factor targeted therapy reactivate tuberculosis? J Rheumatol Suppl. 2005;74:35–9.). [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- However, when the T cell-macrophage balance of the granuloma is disturbed by immune suppression, illness, malnutrition, or old age, organisms emerge and enter a replicative stage, resulting in reactivation of chronic infection, e.g., reactivation tuberculosis.
- A specialized form of immunosuppression has recently been recognized with the current availability and usage of biologic immunomodulators such as anti-cytokine preparations
- The increasing use of anti-TNF-α preparations, for example, for the treatment of rheumatoid arthritis and other inflammatory conditions has unfortunately led to reactivation of chronic infections such as tuberculosis and fungal infections (Figure 7).
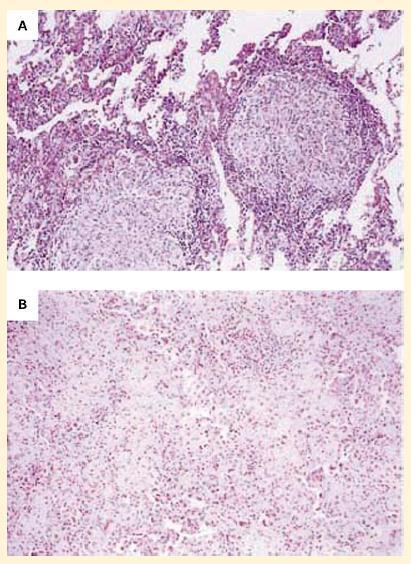
Figure 7. Panel A: Photomicrographs of lung specimens from a patient with tuberculosis and granuloma formation who did not receive infliximab. Panel B: A patient with tuberculosis with a breakdown of granuloma formation who received infliximab. The specimen from the patient who did not receive infliximab shows well-formed granulomas with negligible overt necrosis (panel A). The specimen of lung parenchyma from the patient who received infliximab shows prominent interstitial fibrosis and lymphoid inflammation without granulomas (panel B). (Reproduced with permission from Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–104.) [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Quiz
Related Talks
Henry Mwandumba, Wellcome Trust – TB Immunity in the Lung
Tom Scriba, South African Tuberculosis Vaccine Initiative – TB Immunity
Sara Suliman, South African Tuberculosis Vaccine Initiative – TB Biomarkers



