





- Infectious agents play an aetiologic role in approximately 20% of cancer cases worldwide. At least, more than ten pathogens, including viruses, parasites, and bacteria are known to contribute to oncogenesis either directly via the expression of their protein products or indirectly via chronic inflammation (Table 1).
- To date, there are seven oncogenic viruses [human papillomavirus (HPV), Epstein–Barr virus (EBV), hepatitis virus B and C (HBV and HCV), human T-cell lymphoma virus 1 (HTLV-1), Merkel cell polyomavirus (MCPyV), and Kaposi’s sarcoma virus (KSVH or HHV8)], one oncogenic bacterium (Helicobacter pylori), and three oncogenic parasites (Schistosoma haematobium, Opithorchis viverrini, and Clonorchis sinensis), identified as cancer-related pathogens.
- HBV, HCV, HPV, and H. pylori account for approximately 5% of all cancer cases by leading to hepatocellular carcinoma, cervical cancer and stomach cancer, respectively (Vandeven and Nghiem 2014).
- Pathogens can generally be divided into direct and indirect carcinogens (Figure 1).
- The direct carcinogenic pathogens HPV, HTLV-1, EBV, MCPyV and KSVH share several similarities.
- At least a critical portion of the viral genome can generally be detected in each cancer cell resulting in the expression of viral oncogenes that disrupt cell-cycle checkpoints, inhibit apoptosis and contribute to cell immortalisation.
- In contrast, the indirect carcinogenic pathogens (HBV, HCV, H. pylori, S. haematobium, O. viverrini, and C. sinensis) do not induce expression of oncogenes, but instead their persistent infection leads to a chronic inflammatory state or immunosuppression that limits the anti-tumour immune surveillance mechanisms.
- Because persistent infection is a hallmark of oncogenic pathogens, there is a window of opportunity for cancer prevention by treating the pathogen before malignant progression.
- Viral oncogenic mechanisms generally include: genomic instability, high rates of cell proliferation, resistance to apoptosis, abnormal DNA repair mechanisms, cell polarity changes and interference with telomere shortening, which often coexist with evasion of the antiviral immune response (Morales-Sánchez and Fuentes-Pananá 2014).
- Viral persistence and/or latency, in which there is no or little production of viral particles are biologically compatible with the carcinogenic process, due to avoiding of cell death while maintaining the infectious agent hidden from the immune system.
- Viral persistence in the host is achieved by integrating the viral genome into the cell genome or by expressing viral proteins that equally segregate the viral genome into daughter cells during cell partitioning.
- The net balance between virus and host preserve the integrity of both.
- Cell transformation is probably not an evolutionary viral strategy, but rather a biological accident that rarely occurs in the virus-host interaction.
- All virus-associated tumours result from the cooperation of various events, involving more than persistent infection and viral transformation mechanisms.
- Additional oncogenic hits are necessary for full-blown transformation, and in line with that, an increased mutation rate of infected over normal cells is frequently observed. In this scenario, latently infected cells by oncogenic viruses might be more susceptible targets of additional oncogenic hits (Morales-Sánchez and Fuentes-Pananá, 2014).
- Common mechanisms of indirect carcinogenesis, chronic inflammation, immunosuppression, oncomodulation and chronic antigen-driven lymphoproliferation, are difficult to demonstrate as they cannot be measured by in vitro.
Table 1. Human oncogenic viruses and their associated tumours.
| Human Virus | Associated Tumours |
|---|---|
| EBV | Burkitt’s lymphoma, Hodgkin's lymphoma, immune-suppression-related lymphoma; nasopharyngeal and stomach carcinomas |
| KSHV | Primary effusion lymphoma and Kaposi sarcoma |
| High-risk HPVs | Cervical, head and neck and anogenital tract carcinomas |
| MCPV | Merkel cell carcinoma |
| HBV | Hepatocellular carcinoma |
| HCV | Hepatocellular carcinoma |
| HTLV1 | Adult T-cell leukaemia/lymphoma |
Table 1. [Morales-Sánchez and Fuentes-Pananá, 2014]
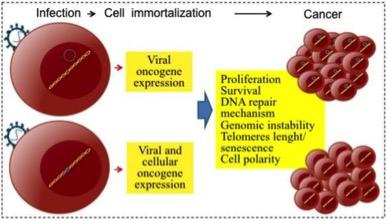
Figure 1. Direct mechanisms of viral carcinogenesis. After infecting target cells, tumour viruses are persistently maintained as genetic elements; viral genomes can form episomes (upper panel example, herpesviruses) or integrate into the host genomic DNA (lower panel example, retroviruses and HBV). [Morales-Sánchez and Fuentes-Pananá, 2014]
Quiz
References and Links
- Morales-Sánchez, Abigail, and Ezequiel M Fuentes-Pananá. 2014. “Human Viruses and Cancer.” Viruses 6 (10). Multidisciplinary Digital Publishing Institute (MDPI): 4047–79.
- Vandeven, Natalie, and Paul Nghiem. 2014. “Pathogen-Driven Cancers and Emerging Immune Therapeutic Strategies.” Cancer Immunology Research 2 (1). NIH Public Access: 9–14.
- Thorley-Lawson, D. EBV Persistence – Introducing the Virus. Curr Top Microbiol Immunol. 390(Pt 1):151-209. (2015)
- Morales-Sánchez, A. & Fuentes-Pananá, E. Human Viruses and Cancer. Viruses. (6): 4047-4079. (2014)
- White, JR., Winter, JA. & Robinson, K. Differential inflammatory response to Helicobacter pylori infection: etiology and clinical outcomes. Journal of Inflammation Research. (8): 137—14. (2015)



