





- Cancer is considered the most common and complex human genetic disease, where a number of molecular networks underlying regulation of cellular fate decisions is perturbed, allowing the uncontrolled proliferation of the damaged cells.
- Cancers figure among the leading causes of morbidity and mortality worldwide, with approximately 14 million new cases and 8.2 million cancer related deaths in 2012.1
- The top five cancer tissues reported in 2012 in men were lung, prostate, colorectum, stomach, and liver, while among women, the most common sites at diagnosis were breast, colorectum, lung, cervix, and stomach.1
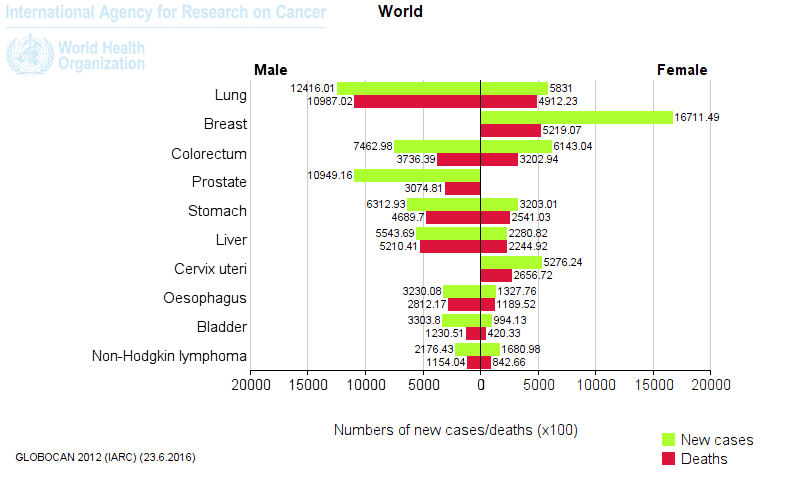
Figure 1. Most common types of cancer in men and women worldwide. [Globocan database 2012]
- Our understanding of the pathobiology of cancer has substantially advanced due to novel theoretical and experimental integrative strategies leading to a more complex, dynamic and interactive network perspective where genetics is in constant dialogue with micro and macro environmental cues that contribute to the aetiology and maintenance of malignant cells (Figure 2).
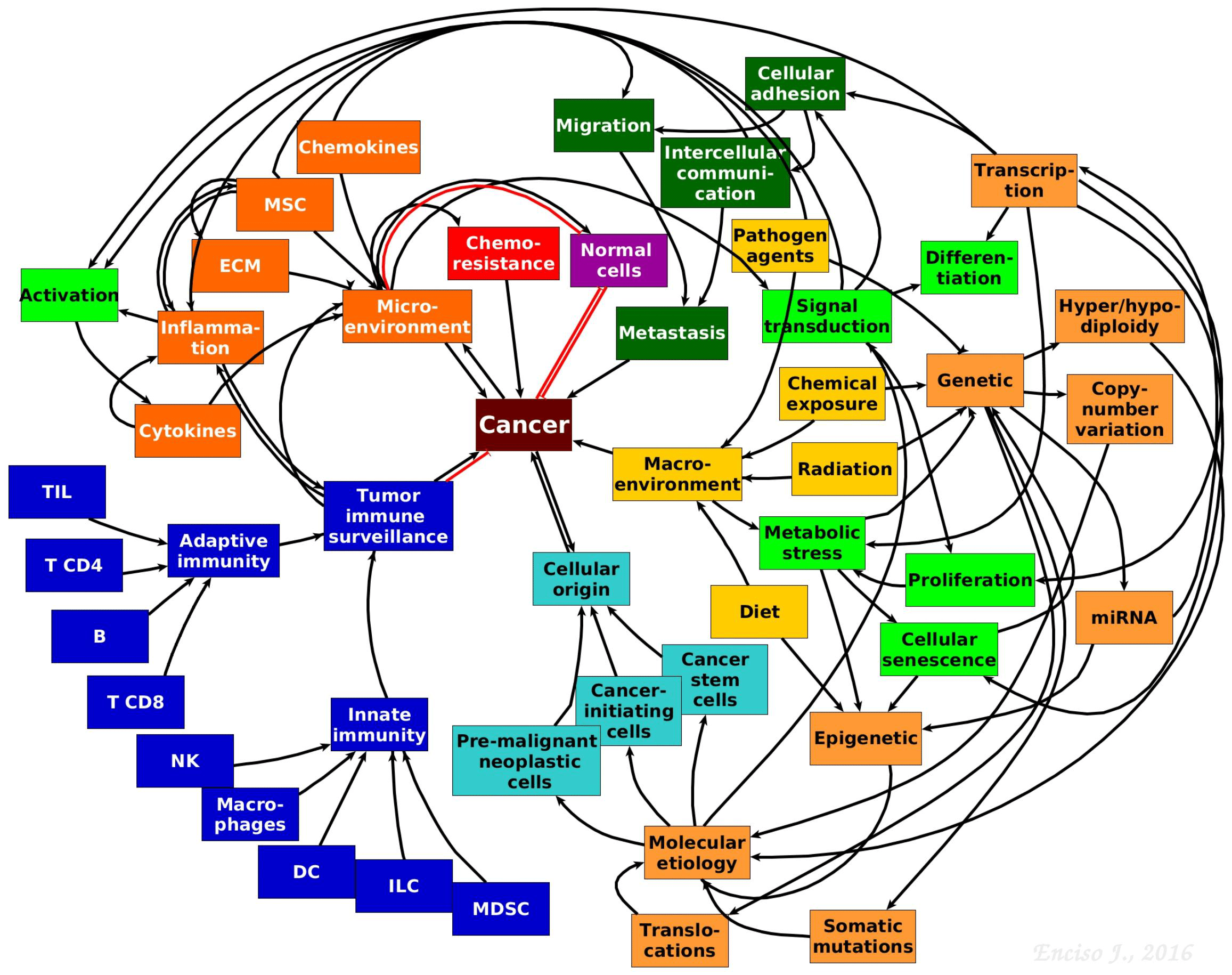
Figure 2. The complexity of tumour biology. Hypothetical model showing the complex and multifactorial origins and progression of cancer. The interactive communication between the immune system, micro- and macro-environmental cues, cancer stem and progenitor cells, cell metabolism, genetics and epigenetics driving tumour progression is depicted. [Jennifer Enciso]
- Changes inducing cell transformation may result from the interaction between genetic factors and microenvironmental cues, that may be in turn influenced by carcinogens and life style.1
- Around one third of cancer deaths are associated to the 5 leading behavioural and dietary risks: tobacco use, alcohol use, high body mass index, low fruit and vegetable intake and lack of physical activity, with the tobacco use being the most important risk factor (near 20%) for global cancer deaths and near 70% of lung cancer deaths.1
- Cancer causing viral infections such as HBV/HCV and HPV are responsible for up to 20% of cancer deaths in low- and middle-income countries.1
- Ageing is another fundamental factor for cancer development. The overall risk accumulation with age is combined with the tendency for cellular repair mechanisms to be less effective (Table 1).1
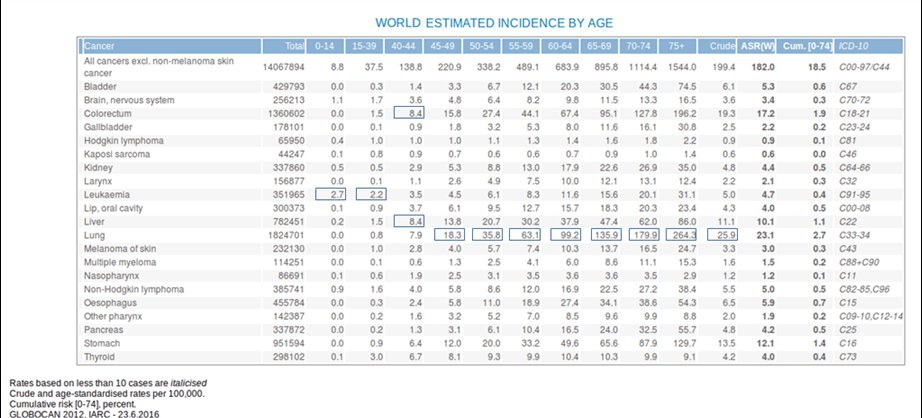
Table 1. Estimated incidence among age groups. [Globocan 2012]
Cancer Genetics
The cell transition from normality to malignancy generally involves either, the inactivation of tumour supressor genes and/or activation of oncogenes.
- The most common tumour suppressor gene mutated among all type of cancers is p53 which encodes a protein involved in cell cycle progression, activation of DNA repair machinery and apoptosis.4
- Frequently mutated genes encode for antigrowth factor receptors (e.g. TGFβ), signal transduction mediators (e.g. Ras proteins, PTEN), cell-cycle regulators (e.g. p16, Rb), supervisors of genome integrity (e.g. Chfr, MLH1, ATM), transcriptional regulators (e.g. VHL, GATA3), adherence mediators principally associated to tumour metastasis (e.g. E-cadherin) and of recent importance, to epigenetic regulators (e.g. DNMT1, MLL3, TET2).4
- Genetic alterations may be spontaneously generated de novo, inherited, macroenvironmentally induced by exposure to carcinogens, virus-induced or associated to ageing-induced genetic instability.
- Many mutations occur as random events in normal cells without generating malignant transformation and are eliminated through immune surveillance. However, some of them generate cancer precursor cells, which in turn are capable of giving rise to cancer initiating cells.
- Common solid tumours (e.g. colon, breast, brain, or pancreas) present somatic mutations in an average of 33 to 66 genes, melanomas and lung tumours ~200 nonsynonymous mutations per tumour, while paediatric tumours and leukemias harbor ~9.6 per tumour.2
- In the 1950’s, Muller and Nording proposed that malignant transformation requires multiple ‘hits’.1 Between two and eight driver gene mutations are found in most tumours.2,4
- Mutations that confer selective growth advantage to tumour cells are called ‘driver mutations’, while mutations induced through cancer progression but conferring no growth advantage are passenger mutations2,3
- To date there are ~140 known genes whose intragenic mutations contribute to cancer, and recent increasing evidence has supported the role of altered epigenetic mechanisms that may also contribute to selective growth advantage.2
- In parallel with point mutations, cancerous cells also present other types of genetic alterations or ‘hits’:
- Hyper/hypodiploidy, e.g. 51-67 chromosomes commonly observed in childhood B-cell precursor acute lymphoblastic leukemia.
- Copy number variations (CNVs), e.g. MCL1 in lung cancer.
- Translocations, e.g. BCR/ABL fusion protein.
- While normal cells are generally induced to apoptosis after DNA damage and chromosome breakage, cancerous cells can survive because of additional mutations that allow them to evade apoptosis.2
- The continuous induction of mutations and clonal selection leads to 4 different types of genetic hetereogenity in cancer:
- Intratumoural: heterogeneity among the cells of the primary tumour.
- Intermetastatic: heterogeneity among different metastatic lesions in the same patient.
- Intrametastatic: heterogeneity among the cells of each metastasis develops as the metastases grow.
- Interpatient: heterogeneity among the tumours of different patients. The mutations in the founder cells of the tumours of these two patients are almost completely distinct.2
Cancer Epigenetics
- Epigenetics, defined as the study of heritable changes in gene expression without alteration in DNA sequences, is also widely targeted in cancer development. Epigenetic regulation considers chemical modifications to the cytosines in the DNA (e.g., methylation, acethylation) or chromatin remodeling (e.g. nucleosomes positioning). 4,5
- The aberrant epigenetic regulation has an important role in human cancer, affecting gene expression patterns and genomic stability mediated by methylation patterns, altered expression of non-coding RNAs, chromosomal looping and, nucleosome positioning and composition.5
- CpG methylation (addition of a methyl group to the 5th carbon of the cytosine ring of a CpG dinucleotide) can contribute to the oncogenic phenotype by general hypomethylation of the cancer genome, by focal hypermethylation at tumour supressor gene promoters or by direct mutagenesis of 5mC-containing sequences by deamination, by UV irradiation, or exposure to other carcinogens.5,6
- Of special interest is the susceptibility of 5-methylcytosine (m5C) residues to spontaneous, carcinogens and light induced mutagenesis.6. Additionally, 5′ cytosine ring methylation increases the formation of carcinogenic adducts induced by chemicals.6
- Nucleosomes, composed by DNA wrapped around a histone octamer, regulate chromatin structure and accessibility to genetic regions in the chromosomes.5 Nucleosome positioning also influence the methylation pattern of cancer-related genes, affecting their transcriptional activation.5
- miRNAs are the most widely type of noncoding RNA studied in normal and cancer cells, regulating ~ 60% of protein-coding genes and have been proposed as biomarkers and prognosis predictors of an increasing number of cancer types.
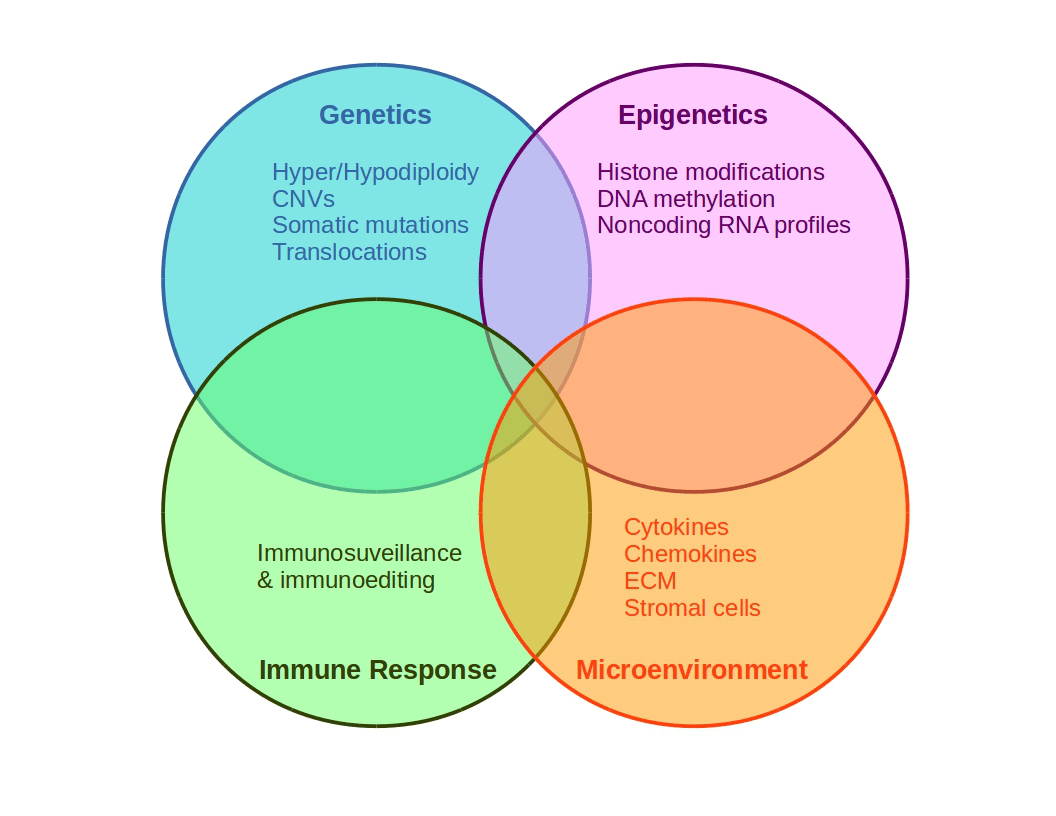
Figure 3. Schematic representation of cancer multifactoriality involving genetic and epigenetic alterations, which may generate favorable conditions for tissue growing.
Quiz
References and Links
- Stewart, B.W. & Wild, C.P. World Cancer Report 2014. Lyon: IARC Press Release 224 (2014).
- Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–58 (2013).
- Knudson, A. G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 1, 157–62 (2001).
- Kandoth, C. et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013).
- Kanwal, R., Gupta, K. & Gupta, S. Cancer epigenetics: an introduction. Methods Mol. Biol. 1238, 3–25 (2015).
- Baylin, S. B. & Jones, P. A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. (2016). doi:10.1101/cshperspect.a019505
- Hayes, J., Peruzzi, P. P. & Lawler, S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol. Med.20,460–9 (2014).
- Leal YA, Fernández-Garrote LM, Mohar-Betancourt A & Meneses-García A. The importance of registries in cancer control. Salud Publica Mex 58:309-316 (2016).
- Villarreal-Garza, C. et al. Real-world outcomes in young women with breast cancer treated with neoadjuvant chemotherapy. Breast Cancer Res Treat. 3811-2 (2016).
- Gomez-Dantes, H. et al. The burden of cancer in Mexico 1990-2013. Salud pública Méx. 58: 118-131 (2016).
- Hanahan, D & Weinberg, R. Hallmarks of Cancer: The Next Generation. Cell. 144: 646–674 (2011).



