





Introduction to Immune Deficiency Disorders
- The immune deficiency conditions are a group of disorders that result from one or more abnormalities of the immune system and that manifest clinically as an increased susceptibility to infection.
- This group of human diseases was ushered in by a single seminal observation made by a clinician who carefully studied a child with recurrent respiratory infections and who later documented the first immune deficiency disorder in the human.
- In 1952, Colonel Ogden Bruton, while searching for the reasons why a young child hospitalized at Walter Reed Army Hospital in Washington, DC, was suffering from repeated and life-threatening infections, found that the child was unable to synthesize specific antibodies and, later, that the child’s serum lacked gamma globulin. Further, the child’s susceptibility to infection was reversed by the administration of serum gamma globulin.
- This landmark discovery represents a consummate example of clinical research that led not only to the current explosive molecular and phenotypic dissection of the immunodeficiencies that are described in this chapter, but that also set the stage for the subsequent development of clinical immunology as we know it today.
General Considerations
- X-linked agammaglobulinemia (XLA) represents the first of over 150 entities that have come to be known as the primary immunodeficiency disorders, some of which will be described here (for a fuller account, refer to Chapter 16 of Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease).
- Although these are rare, they have been useful in unraveling many of the molecular pathways that govern both the innate and the adaptive immune systems and that have provided new diagnostic and therapeutic modalities for the treatment of a wide variety of allergic (Chapter 18, Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease), autoimmune disorders (Chapter 19, Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease), and malignant diseases (Chapters 20 and 21, Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease).
Developmental Background
- Because most of the primary immunodeficiencies result from abnormalities in cellular maturation emanating from known molecular lesions in signaling pathways, transcription molecules, or cytokine systems, it may be useful to frame these defects according to the normal ontogenetic development of the immune system described in sections 1, 2 and 3.
- These are shown schematically in Figure 1.
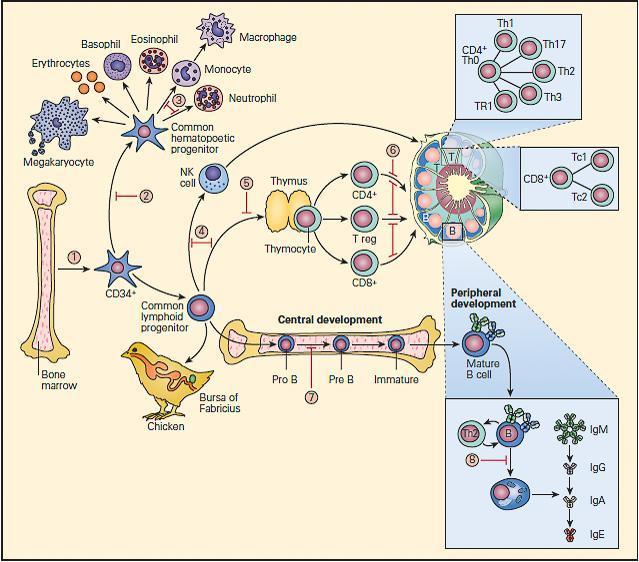
Figure 1. Schematic representation of points in immunologic development at which dysfunction or deficiency can occur in the various primary immunodeficiencies. 1. Reticular dysgenesis; 2. Aplastic anemia; 3. Chronic granulomatous disease; 4. Severe combined immune deficiency (SCID); 5. DiGeorge syndrome; 6. Coronin-1 A deficiency; 7. X-linked agammaglobulinemia (XLA); 8. Common variable immunodeficiency (CVID). [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- The fetal bone marrow provides stem cells that have the potential to progressively differentiate into: (1) a hematopoietic lineage that contains individual precursor cells that give rise to the erythrocytes, granulocytes, monocytes, and platelets; and (2) a lymphopoietic lineage that contains precursor cells that give rise to T lymphocytes, B lymphocytes, and natural killer (NK) cells.
- Mononuclear cells (monocytes) migrate to the lung, liver, spleen, brain, and peripheral lymph nodes where they differentiate into specialized macrophages (e.g., alveolar cells, Kupffer cells, and microglial cells) capable of antigen uptake, processing, and antigen presentation to T lymphocytes.
- Lymphoid precursors are influenced by cytokines as well as a variety of hormonal factors that cause them to differentiate into mature T lymphocytes (e.g., thymic hormones) or B lymphocytes. Although the hormonal source of B cell maturation in the chicken is known to be derived from the bursa of Fabricius, the source of only some of the factors in the bone marrow responsible for B cell differentiation is known in the human.
- T cells may be recognized by the presence of the T cell receptor (TCR) as well as specific membrane proteins referred to as cluster of differentiation (CD) molecules recognized by specific monoclonal antibodies which differentiate lymphocytes into two major families, CD4+ T helper and CD8+ T cytotoxic cells.
- These CD surface proteins on T cells change during maturation. For example, they appear and then disappear as the cell matures from a stem cell to a fully developed T lymphocyte, e.g., double negative CD4−/CD8−, to a double positive CD4+/CD8+, to a specialized mature T cell bearing a single marker, i.e., CD4+ or CD8+ single positive T cells, which emigrate from the thymus into the peripheral tissues.
- B cell differentiation shows similar changes of cell surface proteins with maturation. Pre-B cells have no surface immunoglobulin but later develop surface IgM and IgD immunoglobulins and then lose these as the cell differentiates into fully developed B cells bearing IgM, IgG, IgA, or IgE.
- Until recently, it has been difficult to place the developmental origin of the NK cell in the developmental scheme of monocytes, myeloid cells, T cells, or B cells. NK cells share some surface membrane characteristics of both T lymphocytes and monocytes. It is now recognized that there exists a “natural” population of NK cells and an induced population of NK cells bearing an invariant T cell receptor that are referred to as iNKT cells. These cells also play an important role in killing of cancer cells and viral-infected cells.
The Primary Immunodeficiencies
- The immunodeficiencies have been classically divided into: (1) the phagocytic cell deficiencies, (2) the complement deficiencies, (3) the antibody deficiencies, (4) the cell-mediated deficiencies, and (5) the combined cellular and antibody deficiencies.
- Shown in Figure 2are the relative frequency distributions of the primary immunodeficiencies according to this classification. As can be seen from this figure, the antibody deficiencies are by far the most frequently diagnosed.

Figure 2. Relative distribution of the primary immunodeficiencies according to a more traditional classification. (Adapted with permission from Stiehm ER, editor. Immunologic disorders in infants and children. 5th ed. Philadelphia: Elsevier, Inc.; 2004.) [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].From a comprehensive standpoint, the primary immunodeficiencies may be classified using a more contemporary classification according to the component cell type or transcription molecule or cytokine system they affect (see Tables 16A-1to 16A-8 in chapter 16 of Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012).
Defects of Oxidative Metabolism
Chronic Granulomatous Disease (CGD) (MIM ID #306400)
- This disorder is the most common of the syndromes associated with defective oxidative metabolism resulting in diminished bactericidal activity. In most cases, it is inherited as an X-linked trait, although autosomal recessive forms are known.
- The underlying defect is impaired generation of activated forms of oxygen, i.e., superoxide (O2−) and hydrogen peroxide (H2O2) due to a variety of enzymatic defects involving the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system
- The NADPH oxidase is a multicomponent enzyme complex required for the generation of superoxide and its metabolites hydrogen peroxide and bleach. The structural components are referred to as phox (phagocyte oxidase) proteins.
- At rest, the complex exists as separate components: the cytochrome b558 is comprised of gp91phox, the 91kd beta chain, and p22phoxthe 22kd alpha chain. Together they form a complex that binds heme and flavin, embedded in the walls of secondary granules.
- In the cytosol are the structural proteins p47phoxand p67phox, and the regulatory components p40phox and RAC. p47phox and p67phox are phosphorylated and bind tightly together on neutrophil activation.
- In association with p40phoxand RAC, they join to the complex of gp91phox and p22phox to form the intact NADPH oxidase. This complex harvests an electron from NADPH and donates it to molecular oxygen, creating superoxide (O2−). Superoxide dismutase (SOD) converts O2− to H2O2. Myeloperoxidase converts H2O2 to bleach by combination with chlorine. Phagocyte production of O2− facilitates activation of certain proteins inside the phagosome.
- Mutations in gp91phox, p22phox, p47phox, and p67phoxcause chronic granulomatous disease (CGD), characterized by recurrent life-threatening infections due to catalase-positive bacteria and fungi and granulomatous complications (MIM #306400, 233690, 233700, and 233710).
- Shown in Figure 3 are the NADPH oxidase mutations that give rise to CGD together with their inheritance patterns, chromosomal locations, and relative frequencies.
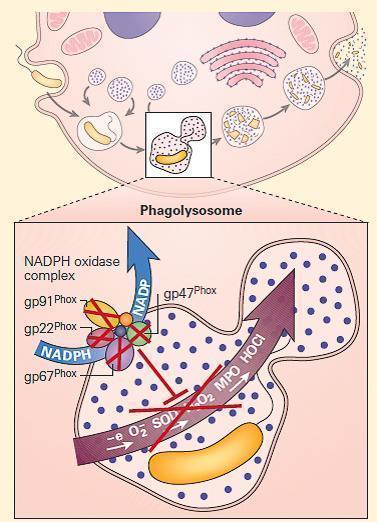
Figure 3. Schematic representation of a neutrophil in chronic granulomatous disease (CGD). The NADPH oxidase is assembled on either the wall of the phagolysosome, or the plasma membrane, depending on whether the invader has been engulfed or is too large to be ingested. After cellular activation the secondary granule fuses with the phagolysosome, embedding the cytochrome components in the wall of the vesicle. p47phox and p67phox become phosphorylated and tightly associated in the cytosol, followed by transit to the transmembrane cytochrome complex. When p40phox and RAC join the complex, the mature NADPH oxidase is formed, and is capable of harvesting an electron from NADPH, oxidizing it to NADP+ and delivers that electron to molecular oxygen in the phagosome, generating superoxide (O2-). Superoxide is converted to hydrogen peroxide (H2O2) by superoxide dismutase (SOD); this in turn is converted to hypohalous acid (HOCl or bleach) by myeloperoxidase (MPO). Mutations in the four structural genes of the NADPH oxidase (gp91phox, p22phox, p47phox, and p67phox) cause CGD. Gp91phox is X-linked recessive, while the others are autosomal recessive. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- The gp91phoxvariant is X-linked, located on Xp21, and accounts for about two-thirds of cases; the other three variants have an autosomal recessive pattern of transmission, and p47phox, located on chromosome 7, accounts for about 25 percent, with p22phox, located on chromosome 16, and p67phox, located on chromosome 1q42, accounting for the rest.
- There are no autosomal dominant cases of CGD. CGD occurs in 1/200,000 live births, but this may be an underestimate. The majority of patients are diagnosed as toddlers and young children; infections or granulomatous lesions are usually the first manifestations.
- Symptoms usually begin during the first few years of life, with the advent of recurrent disseminated abscesses or pneumonias.
- The characteristic suppurative granulomas scattered throughout the body at these sites gave rise to the name of the entity. Individual granulomas consist of peripheral collections of lymphocytes and macrophages surrounding a central necrotic core, superficially resembling the granulomas seen in tuberculosis.
- The epidemiology of CGD infections in less developed countries is not as well defined, but it differs slightly from North America and Europe.
- In countries where BCG vaccination is still administered, local BCG lymphadenitis is common.
- In North America, the overwhelming majority of infections in CGD are due to only five organisms: aureus, Burkholderia cepaciacomplex, Serratia marcescens, Nocardia, and Aspergillus.
- In contrast to other primary immunodeficiencies, the infectious agents found in CGD are of low virulence in normal individuals and are characteristically catalase-positive.
- Certain catalase-negative organisms, such as Streptococcus pneumoniaeand Streptococcus pyogenes, which also produce hydrogen peroxide, do not often infect these patients.
- Trimethoprim/sulfamethoxazole (TMP/SMX) prophylaxis has reduced the frequency of bacterial infections markedly. In patients receiving TMP/SMX prophylaxis, staphylococcal infections are essentially confined to the liver and cervical lymph nodes.
- In recent years, fungal infections, typically those due to Aspergillusspecies, have become more predominant. Although itraconazole prophylaxis has been shown to reduce fungal infection, newer antifungals, such as voriconazole and posaconazole, should further reduce fungal mortality in CGD.
- The use of IFN-γ has been shown to reduce the number and severity of infections in CGD by 70 percent compared to placebos. Therefore, current recommended prophylaxis in CGD is trimethoprim/sulfamethoxazole, itraconazole, and interferon gamma.
- The gastrointestinal and genitourinary tracts are frequently affected by granulomata. Esophageal, jejunal, ileal, cecal, rectal, and perirectal involvement with granulomata mimicking Crohn’s disease have been described, and affect 43 percent of patients with X-linked CGD and 11 percent of those with p47phox
- Gastric outlet obstruction is common and may be the initial presentation of CGD. Bladder granulomata and ureteral obstruction are common in patients with defects in gp91phoxand p22phox and readily relieved with steroids.
- Prednisone 1 mg/kg for a brief initial period then tapered to a low dose on alternate days is usually successful.
- However, relapse or recurrence of gastrointestinal granulomatous disease is common, requiring the frequent use of prolonged low-dose steroid therapy. The use of infliximab increases the rates of fungal and bacterial infection in CGD, just as it does in normal individuals.
- The X-linked gp91phoxcarrier females typically have two populations of phagocytes: one that produces superoxide and one that does not. Discoid lupus erythematosus-like lesions, aphthous oral ulcers, and photosensitive rashes are seen in gp91phox Infections are not usually seen in female carriers unless the population of normal neutrophils is below 5 to 10 percent; then, these carriers are at risk for CGD type infections.
- The diagnosis of CGD is made by demonstration of reduced or absent superoxide generation.
- Although the nitroblue-tetrazolium (NBT) assay was the most widely known diagnostic test for chronic granulomatous disease in the past, which was based on the direct reduction of NBT by superoxide free radical to form an insoluble blue formazan in the normal and its absence in the patient, it has been largely replaced by the flow cytometric dihydrorhodamine (DHR) assay, which is now preferred because of its speed, ease of use, ability to distinguish X-linked from autosomal patterns of CGD, and its sensitivity to very low numbers of functional neutrophils.
- Immunoblot and mutation analysis has also been used to identify specific proteins and mutations. The precise gene defect should be determined when possible, as it is critical for genetic counseling and is prognostically significant.
- p47phoxdeficiency has a significantly better prognosis than X-linked disease: mortality for the X-linked form has been shown to be about 5 percent per year, compared to 2 percent per year for the autosomal recessive varieties.
- Bone marrow transplantation leading to stable chimerism has been successfully performed in patients with CGD, including for refractory infection, predominantly from Aspergillus.
- In some studies using reduced-intensity non-ablative bone marrow transplantation from HLA-identical siblings into CGD patients, success was greater in children than adults, but transplant-related toxicities, such as graft versus host disease, remained problematic.
- Gene therapy for p47phoxand gp91phox deficiencies have been successful, but not durable.
The Leukocyte Adhesion Deficiencies
- Over the years, a number of defects of leukocyte movement from the blood into tissues has been identified which predispose patients to recurrent infection.
- These include a group of molecular defects that come under the general heading of leukocyte adhesion deficiency (LAD).
- These molecular lesions are responsible for the clinical manifestations resulting from an impaired step in the inflammatory process, namely, the emigration of leukocytes from the blood vessels to sites of infection, which requires adhesion of leukocytes to the endothelium.
- Leukocyte adhesion to each other, endothelium, and to bacteria is required for travel, communication, and inflammation to fight infection. The leukocyte adhesion molecules, predominantly the integrins and selectins, mediate these processes.
- Leukocyte β2 integrins are heterodimeric molecules on neutrophils, monocytes, and lymphocytes that attach to intercellular adhesion molecules (ICAMs) on the endothelium in order to exit the circulation (Figure 4). ICAMs are also expressed on other leukocytes, mediating some forms of cell-cell adhesion. Certain β2 integrins bind directly to pathogens or to a complement.
- The integrins are composed of an α chain (CD11a, CD11b, or CD11c) noncovalently linked to a common β2 subunit, CD18.
- The αβ heterodimers of the β2 integrin family are CD11a/CD18 (lymphocyte-function-associated antigen-1, [LFA-1]), CD11b/CD18 (macrophage antigen-1, [Maca-1]; complement receptor-3, [CR3]), and CD11c/CD18 (p150,95; complement receptor-4, [CR4]).
- Since CD18 is required for normal expression of the αβ heterodimers, mutations that eliminate or impair CD18 lead to either very low or no expression of CD11a, CD11b, and/or CD11c.
- These mutations lead to inability of leukocytes to bind to endothelium, with each other, to certain pathogens, or to complement opsonized particles, thereby causing leukocyte adhesion deficiency type I (ITGB2, 21q22.3; MIM #116920).
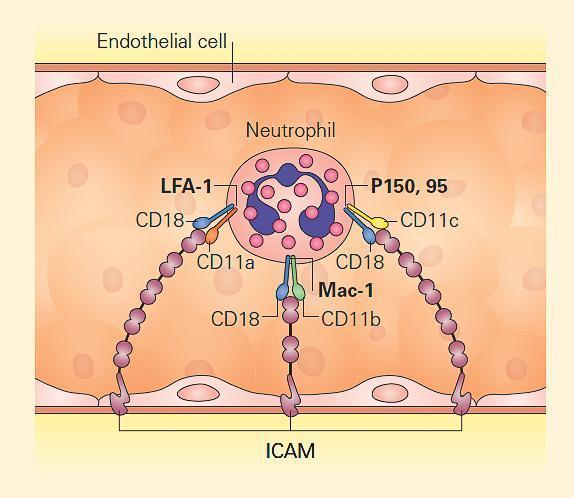
Figure 4. Leukocyte adhesion via integrins. Neutrophils attach to endothelium via PMN-surface receptors composed of heterodimeric CD18 molecules with their partners, CD11a, CD11b, and CD11c. These in turn bind to cell surface molecules on the endothelium, composed of the intercellular adhesion molecules (ICAM) 1 and 2. Mutation of CD18 leads to loss or impairment of all the heterodimers, disabling tight adhesion and vascular exit of neutrophils. Therefore, CD18 deficiency leads to leukocyte adhesion deficiency type I (LAD I). [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- In addition to LAD I, two additional defects of the leukocyte adhesion cascade have been described, referred to as LAD II and LAD III, involving several precise ordered steps such as rolling, integrin activation, and firm adhesion of the leukocytes (for a fuller description, look at the resource section below and Chapter 16 of Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012).
Defects of the Innate Immune System
- One of the most rapidly developing areas of immunologic research that is finding clinical applicability is being directed to the study of genetic mutations involving signaling pathways and pattern recognition receptors (PRRs) involved in innate immune function, e.g., TLRs that are associated with severe innate immunodeficiency phenotypes (section 1).
- These present powerful opportunities to determine the relationship between specific immunological defects and human disease processes in vivo.
- There are several emerging studies of human primary immunodeficiencies associated with abnormal TLR signaling that demonstrate that this pathway is critical for human defense against infection.
- TLRs mediate recognition of microbes, regulate activation of the innate immune response, and provide a linkage with adaptive immune responses.
- Cellular and molecular studies over the past several years have identified a number of common TLR polymorphisms that modify the cellular immune response and production of cytokines in vitro. In addition, human genetic studies suggest that some of these polymorphisms are associated with susceptibility to a spectrum of diseases, particularly the infectious diseases.
- Signaling pathways activated through receptors for IL-1β, IL-18, TNF-α, CD40, and for many of the TLRs are shared with those for ectoderm formation.
- These pathways converge at the activation of NF-κB, an important transcription factor dependent on the activation of the inhibitor of the NF-κB kinase (IKK) complex and its subsequent phosphorylation of the NF-κB inhibitor, IκB (Chapter 9 in Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012).
- Defects in one of the IKK components, IKKγ, also called the NF-κB essential modulator (NEMO), cause ectodermal dysplasia together with a complex set of immunodeficiencies with dysfunction of the innate (Toll-like receptors, TNF-αR, and IL-1R) and adaptive (CD40 and IL-18) immune systems (Figure 5).
- These patients have a very significant susceptibility to nontuberculous mycobacterial infections, which may be mediated through IL-12 induction.
- These patients may also require prophylactic antibiotics and intravenous immunoglobulin due to ineffective immunoglobulin class switching (Chapter 6 of Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012 and Immunopaedia case studies: An 8-year-old boy with recurrent respiratory infections & Immunodeficiency and failure to thrive).
- Some patients respond well to IFN-γ treatment in the setting of disseminated mycobacterial infection. This syndrome is described in greater detail below together with the hyper IgM syndromes.
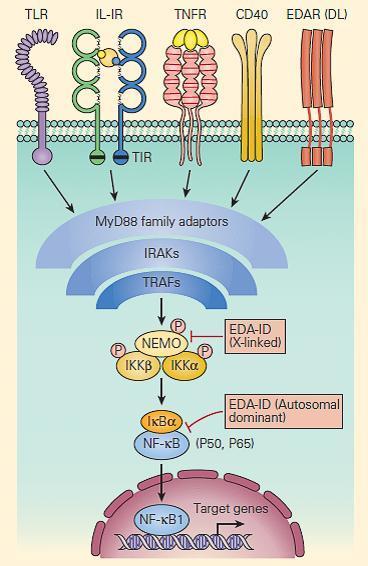
Figure 5. Schematic representation of NF-kB activation and NEMO deficiency. Signaling pathways, including those for ectodermal formation (EDA/DL) and inflammation (TNFR, IL1R, IL18R, CD40R, and TLR), all converge on the pathway needed to phosphorylate and ubiquitinate the inhibitor of NF-kB, i.e., IkBα. When this occurs appropriately it leads to liberation of the active p50/p65 NF-kB, which, after translocation to the nucleus, activates proinflammatory genes as well as genes responsible for skin and skeletal morphogenesis. Therefore, mutations in NEMO have the potential to impair both ectoderm formation and innate immunity. However, defective CD40 signaling also leads to impaired immunoglobulin class-switch recombination, in some cases, causing failure to switch from immunoglobulin M (IgM) to IgG, hence theoccurrence of hyper IgM in some patients. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Lymphocyte Immune Deficiencies
Combined T Cell and B Cell Defects
- T lymphocytes are the pivotal cells sine qua nonfor orchestration of adaptive immunity.
- They direct the killing of intracellular pathogens and the responses of B lymphocytes and antigen-presenting cells (APCs) in the defense against extracellular pathogens (sections 1 & 2).
- They can also kill directly when they recognize aberrant surface molecules.
- Consequently, defects in T cell development and/or function invariably can also result in defects affecting B cell, NK cell, and myeloid compartments.
- For ease of discussion, these have been grouped both according to their functional sites of derangement, e.g., signaling, DNA rearrangement and repair or purine metabolic sites, as well as their phenotypic severity, e.g., SCID versus non-SCID.
- Figure 6 shows a schematic representation of these combined T and B cell defects portrayed according to this arbitrary classification.
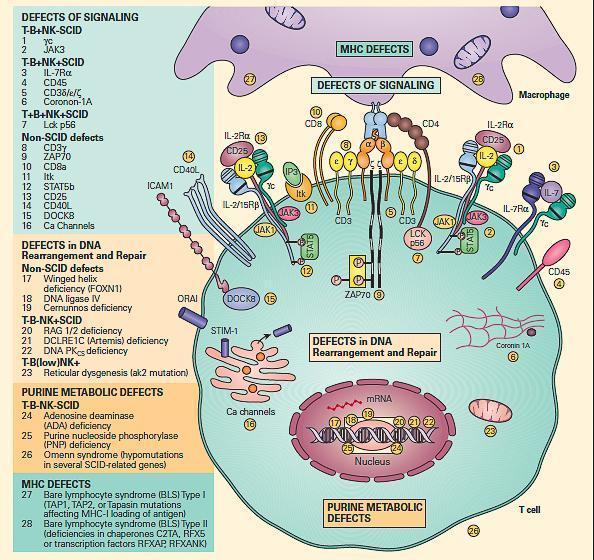
Figure 6. Schematic representation of the different levels at which defects of the combined T and B cell deficiencies occur divided according to the arbitrary classification. These include defects of signaling, defects in DNA rearrangement and repair, and purine metabolic defects. Shown on the right side of the figure are molecular defects seen in conditions presenting as the SCID phenotype arranged according to various combinations of involvement of T, B, and NK phenotypes. On the left side of the figure are shown other molecular defects seen in non-SCID phenotypes. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Severe Combined Immune Deficiencies (SCID)
- Severe combined immunodeficiency (SCID) is a collection of severe heritable genetic immunodeficiency disorders in which both “arms” (B cells and T cells) of the adaptive immune system may be crippled due to a defect in one of several possible genes.
- It is known as the “bubble boy” disease, named after David Vetter who survived isolated in a plastic bubble for 12 years because of the condition’s extreme vulnerability to infectious diseases.
- The overall incidence is estimated to be 1 in 75,000 births, and without treatment, SCID patients usually die during infancy.
- Several different forms of SCID have been described based on differential involvement of T, B, and NK cell lineages. There are several types of SCID that are characterized by defective T cell function with different levels of B cell and NK cell impairment (Figure 6).
- When considering the diagnosis of SCID, the most serious of the immune deficiency disorders, it is important to keep several things in mind.
- SCID is a medical emergency. If the patient with SCID is to survive transplantation, it is important to avoid community viral infections, which can be devastating;
- Persistent engraftment of maternal lymphocytes, i.e., graft-versus-host disease (GVHD), is both a symptom of SCID and a cause of many of the symptoms, such as failure to thrive and rash;
- A phenotype of infections and clinical illness characterize the presentation of these patients, and the focus of laboratory testing is directed at efforts to confirm the genetic defects;
- Since some mutations in patients with SCID are of the missense type, proteins can still be produced and detected by flow cytometry but are non-functional. Therefore, the clinical picture, typically developed out of the infection profile and the flow cytometry information, must ultimately guide decisions about which genes to sequence.
The Clinical Presentation of Patients with SCID
- SCID typically presents early in life with failure to thrive, diarrhea, recurrent infections due to respiratory viruses, herpes viruses, Pneumocystis (carinii) jiroveci, and bacteria.
- Although mucosal candidiasis is common, aspergillosis is not.
- In contrast to patients with interferon-γ/IL-12 pathway defects, described previously, where susceptibility to both mycobacteria and BCG is present, generalized complications of BCG vaccine are common in patients with SCID when the vaccine is administered, but nontuberculous mycobacterial infections are not.
- The underlying defects are caused by improper lymphocyte proliferation due to failure of signal transduction (i.e., receptor defects) or toxicity (adenosine accumulation and lymphocyte death).
- Therefore, absolute lymphopenia is a common finding in many forms of SCID.
- In addition, T lymphocyte failure may facilitate persistent engraftment of maternal T lymphocytes transferred to the fetus during pregnancy, leading subsequently to the alloreactive response of the engrafted maternal T cells against the fetus referred to as GVHD, which typically presents in the neonatal infant as rash and/or diarrhea.
- Treatment for all forms of SCID should include prevention of viral infection, prophylaxis against Pneumocystis jiroveciwith trimethoprim-sulfamethoxazole, and consideration of early bone marrow transplantation or gene correction. The overall frequency of SCID is approximately 1/50,000
Defects of Cytokine and Signaling
IL-2 Receptor Common Gamma Chain Deficiency T- B+ NK- SCID (MIM ID #300400)
- The prototype for the most common form of SCID has been considered to be the X-linked (SCID-XL)form due to mutations in the shared IL-2 common gamma chain receptor (IL-2Rγc, Xq13) found in receptors for IL-2, IL-15, IL-4, IL-7, IL-9, and IL-21 (Figure 6).
- Figure 7 shows the central role which the IL-2Rγc chain plays in immune responsiveness since this component is an obligatory signal transducing chain for several cytokine receptors, including IL-2R, IL-15R, IL-4R, IL-7R, IL-9R, and IL-21R (hence the designation common gamma chain).
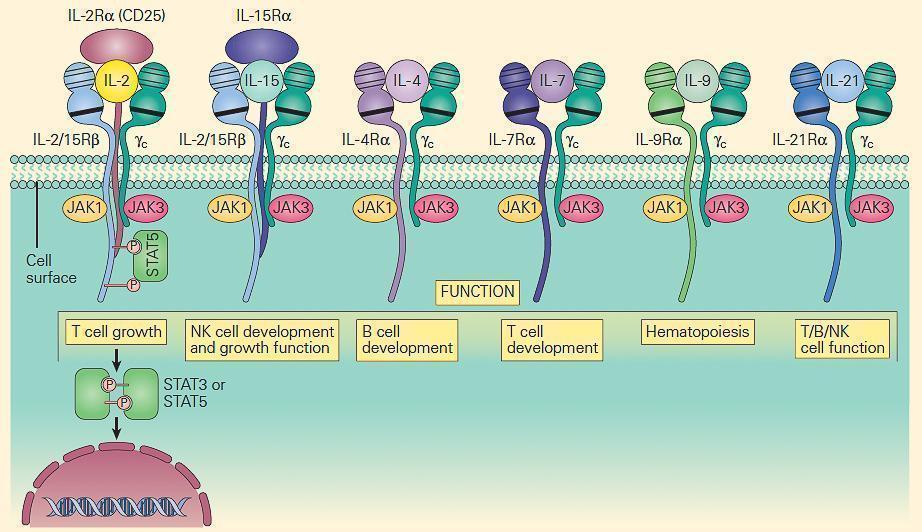
Figure 7. Schematic representation of the receptors that use the IL-2R common gamma chain (γc). Note that γc is linked to JAK3, and these receptors signal through STAT3 and STAT5. Therefore, mutations in γc, JAK3, and STAT5 have many overlapping features. Γc mutations exert part of their profound effect through their impact on receptors other than IL-2. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Defects in B Cells
- B cells begin their antigen-independent life in the bone marrow, but it is only in peripheral lymphoid tissues during the antigen-dependent phase where they encounter antigen, experience class-switch recombination (CSR) and expansion, and become the predominant cells in these tissues (Chapters 2and 6 of Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012]).
- Their ontogeny and differentiation have been largely defined through the arduous characterization of immune defects leading to hypogammaglobulinemia, beginning with the molecular study of the flagship entity, X-linked agammaglobulinemia.
- The major B cell defects can be divided into three major categories of antibody deficiencies:
- (1) defects in early B cell development,
- (2) hyper-IgM syndromes (also called class-switch recombination defects), and
- (3) common variable immunodeficiency (CVID) (Figure 8).
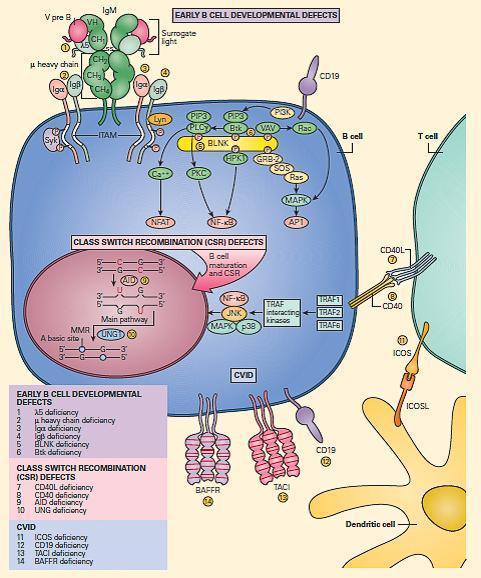
Figure 8. Schematic representation of the molecular events that are involved in B cell signaling. Primary B cell signaling occurs, through the BCR (IgM)-associated signaling molecules Igα/β, as well as the co-stimulatory molecules CD21 and CD19. Proximal signaling occurs through Syk, BTK, BLNK, Lyn, PLCg, and PKC. Downstream effector molecules include nuclear factor kB (NF-kB) and NFAT that translocate to the nucleus, where they affect gene transcription that controls B cell proliferation, survival, differentiation, and migration. The known human signaling defects resulting in primary immune deficiencies of B cells include (1) mutation in BTK, (2) m heavy chain deficiency (IgM), (3) l5 deficiency, (4) Igα deficiency, (5) Igβ deficiency, and (6) BLNK deficiency. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
Defects in Early B Cell Development
B Cell Defects with Severe Reduction in All Serum Ig Isotypes with Absent B Cells: X-Linked Agammaglobulinemia (XLA) (MIM ID #300755)
- First described in 1952 by Bruton, severe X-linked agammaglobulinemia (XLA) has been the paradigm for understanding the pure role of the B cell in immune function.
- The disease is now known to be due to defects in a tyrosine kinase, Bruton tyrosine kinase, BTK (MIM #300300).
- Since BTK mutations lead to severe defects in B cell development past the pro-B cell stage, circulating B cells are typically absent altogether in XLA in contrast to other B cell defects where these cells may be present.
- Because XLA affects almost exclusively the ability to produce antibody, it is not usually clinically apparent until after transplacental maternal antibody has waned after six months of age.
- Typical presentations include upper and lower respiratory infections, diarrhea, cellulitis, meningitis, and sepsis.
- Infections are typically caused by Gram-positive, highly pathogenic encapsulated organisms such as Streptococcus pneumoniaand Haemophilus influenzae type b.
- Infections caused by Gram-negative organisms, including Campylobacterspecies and Pseudomonas as well as other organisms such as Pneumocystis and herpes viruses, are not common.
- Although susceptibility to infection with high-grade encapsulated bacteria has been the hallmark presentation of patients with XLA, it is also known that these patients can also suffer from certain viral infections.
- Enterovirus infections have been especially severe in XLA, often causing a severe meningoencephalitis. The basis for this susceptibility of XLA patients to viral infection recently has been shown to be due to a special role of BTK as a key signaling molecule that interacts with and acts downstream of TLR8 and TLR9 in signal transduction, making for a unique innate defect in addition to antibody deficiency
Defects in Class-Switch Recombination (CSR)
Severe Reduction in Serum IgG and IgA with Normal/Elevated IgM and Normal Numbers of B Cells: The Hyper IgM Syndromes
- This is a second major class of immunodeficiencies with relatively normal numbers of hypofunctional T, B, and NK cells characterized by infections similar to those seen in patients with SCID, but typically with a more delayed and slightly milder presentation.
- Because of the fundamental T cell functional defects in these diseases, without defects in cellular survival, the suspicion of an immune deficiency may come less from the initial laboratory examination and more from careful piecing together of the clinical history, infection profile, and functional studies.
- Some of these cases have normal to increased IgM with low IgA and IgG and normal B cell numbers. There are four syndromes in this category recognized so far, and they are sometimes collectively grouped as the hyper IgM syndromes. These include
- (1) CD40 ligand (CD154) deficiency;
- (2) CD40 deficiency;
- (3) activation-induced cytidine deaminase deficiency (AID); and
- (4) uracil DNA glycosylase (UNG) deficiency.
CD40 Ligand (CD154) Deficiency (MIM ID #308230)
- CD40L is a T cell membrane protein that interacts with CD40, its cognate receptor, on B cells, dendritic cells, monocytes, macrophages, and activated epithelial cells.
- CD40L on T cells activates CD40 to drive B cell immunoglobulin class switch from IgM to IgG (class-switch recombination, CSR), as well as for subsequent maturation of antibody and formation of normal lymph node anatomy.
- Figure 9 shows a schematic representation of the critical role of CD40L/CD40 binding for the class switch and shows how a deficiency of the CD40 can lead to partial IgG/IgA/IgE hypogammaglobulinemia with normal or elevated IgM levels.
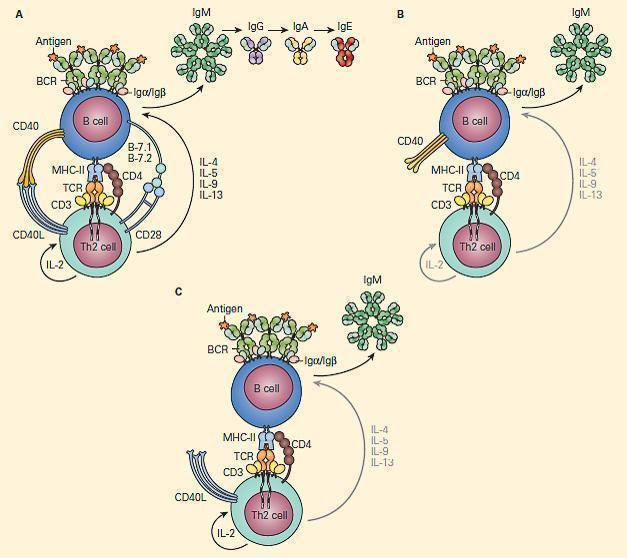
Figure 9. Schematic representation of the critical interaction of CD154 (CD40L) expression on the T cell with the CD40 on the B cell that is required for IgM/IgG class switching. Panel A: After cognate interaction of the MHC-II/peptide complex with the TCR of a CD4+T cell, CD40L activates CD40 to drive B cell immunoglobulin class switch from IgM to IgG (class-switch recombination, CSR), as well as subsequent maturation of antibody from IgM→IgG→IgA→IgE. Panel B: In the absence of CD40L, there is no CD40L/CD40 interaction, no class switch, and there occurs only synthesis of IgM. Panel C: Similarly, in the absence of CD40 and lack of binding with CD40L, there is no class switch and synthesis only of IgM. [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- In addition to directing B cell function, CD40L is critical for the control of certain intracellular parasites that are characteristic of SCID, such as Pneumocystis jiroveci.
- Other characteristic infections include Cryptosporidium, with eventual hepatic involvement, leading to sclerosing cholangitis, and histoplasmosis.
- Liver damage significantly reduces the success of bone marrow transplantation, and therefore filtered water that is free of Cryptosporidiumis recommended once the diagnosis is suspected (this precaution may or may not be necessary, depending on regional water supplies).
- Other significant complications of CD40L deficiency include neutropenia, which may respond to G-CSF or IVIG, and malignancy, which may develop in adolescence or adulthood.
- See Immunopaedia Case study: An 8 year old boy with recurrent respiratory infections
CD40 Deficiency (MIM ID #109535)
- Since CD40 is the cognate receptor for CD40L, it is expected that mutations in CD40 should resemble CD40L defects at both clinical and laboratory levels.
- This is indeed the case, and patients with CD40 deficiency present with hypogammaglobulinemia, impaired antibody maturation, and impaired lymphoid organ development and anatomy.
- The few patients identified have had Pneumocystis, Pseudomonas, and Cryptosporidiuminfections, all with clinical characteristics similar to patients with CD40L deficiency.
Quiz



