





Introduction
- Most autoimmune diseases are characterized by the production of antibodies directed to self-antigens.
- It is important to determine whether the presence of an autoantibody is causally related to the pathogenesis of disease or represents an autoimmune epiphenomenon playing no causal role in its development.
- The physical properties of an autoantibody that determines its pathogenicity include:
- its affinity to the antigen that plays a role in immune complexes formation
- its charge that helps antibody attach to tissues.
- In SLE, positively charged autoantibodies (anti-double stranded DNA) associate with the negatively charged basement membrane in the kidney, where they can form complexes in situ with DNA and lead to local inflammation.
- Clinically Breg are implicated in inflammation and autoimmunity (such as in animal models of systemic lupus erythematosus).
- However, most studies on Breg have been done in vitro or in murine models; less is known about Breg functions in healthy humans and human autoimmune diseases.
- Although autoimmune patients display an increased Breg frequency, Breg from SLE patients have impaired suppressive activity due to a defect in IL-10 production.
Systemic lupus erythematosus (SLE)
- SLE is a prototypic autoimmune disease characterized by multi-organ inflammation, with a diverse array of clinical manifestations and production of pathogenic autoantibodies directed against nucleic acids, reflecting a global loss of self-tolerance.
- The exact patho-aetiology of SLE remains elusive. The loss of tolerance with subsequent immune dysregulation is a consequence of an extremely complicated and multifactorial interaction among various genetic factors implicating over 30 genetic loci in the setting of environmental triggers and stochastic events.
- Defective immune regulatory mechanisms, such as the clearance of apoptotic cells and immune complexes, are important contributors to the development of SLE. The loss of immune tolerance, increased antigenic load, excess T cell help, defective B cell suppression, and shifting of T helper 1 (Th1) to Th2 immune responses lead to B cell hyperactivity and the production of pathogenic autoantibodies.
- Aberrant innate immune responses play a significant role in the pathogenesis of SLE, contributing both to tissue injury via release of inflammatory cytokines and to aberrant activation of autoreactive T and B cells, with the latter leading to pathogenic autoantibody production and resulting in end-organ injury.
- The basic pathological features of SLE are inflammation and blood vessel abnormalities, which include band or occlusive vasculopathy, vasculitis, and immune complex deposition. The most characterized organ pathology is in the kidney. By light and immunofluorescence microscopy, renal biopsies in patients with SLE display mesangial cell proliferation, inflammation, basement membrane abnormalities, and immune complex deposition (immunoglobulins and complement components).
- The central immunological disturbance in patients with SLE is autoantibody production. These antibodies are directed at several self-molecules found in the nucleus, cytoplasm and cell surface, in addition to soluble molecules such as IgG and coagulation factors. Anti-nuclear antibodies are most characteristic and present in more than 95% of patients. Anti-double stranded DNA (ds-DNA) and anti-Sm antibodies are unique to patients with SLE. In fact, their presence is included in the classification criteria of SLE. Anti-DNA antibody titers frequently vary over time and disease activity while anti-Sm antibody titers are usually constant.
The waste disposal Hypothesis and SLE
- It is now clearly established that complement can have both pro- and anti-inflammatory activities.
- Normal functioning of the complement system may actually reduce inflammation by promoting prompt killing and removal of infecting organisms, solubilizing antigen-antibody precipitates, clearing immune complexes from the circulation, and promoting disposal of apoptotic cells and debris.
- Normal cells constantly undergo apoptosis and must be removed and digested without being presented as antigens and evoking harmful immunologic responses.
- If this normal clearing mechanism does not occur efficiently, intracellular components can accumulate and initiate autoimmune responses.
- This may explain the very high frequency of systemic lupus erythematosus (SLE) in patients with early complement component deficiencies.
- FIGURE 1 shows three variations of the waste-disposal hypothesis together with consequences of its failure.
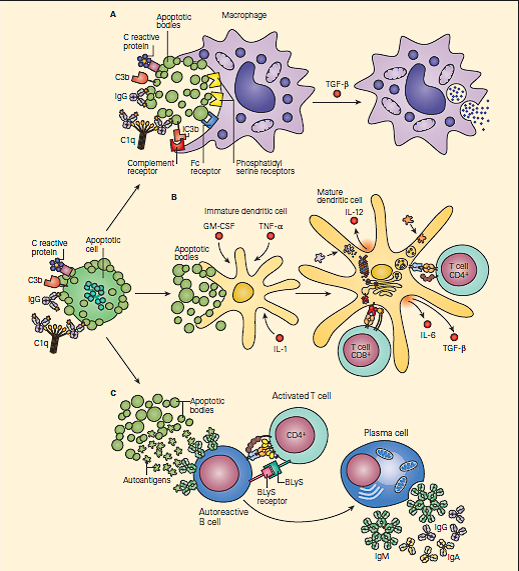
Figur 1: The waste-disposal hypothesis for systemic lupus erythematosus. Panel A: A macrophage is shown engulfing apoptotic bodies released from an apoptotic cell. There are a variety of ligands on apoptotic cells and receptors on macrophages that make this process extremely efficient. The binding of C1q, C-reactive protein, and IgG to apoptotic cells may promote the activation of complement, leading to the clearance of apoptotic cells by ligation of complement receptors. Once the macrophage has engulfed the apoptotic cell, it secretes the anti-inflammatory cytokine transforming growth factor β (TGF-β). Panel B: When there is an excess of apoptotic cells and the failure of one or more of the normal systems of receptor-ligand recognition for the uptake of apoptotic cells, immature dendritic cells may take up apoptotic bodies. If this occurs in the presence of inflammatory cytokines such as granulocyte-macrophage colony-stimulating factor (GM-CSF), tumor necrosis factorα (TNF-α), and interleukin-1, the dendritic cell may mature into an autoantigen-presenting cell. The dendritic cell is shown presenting autoantigens to a T cell in the presence of costimulatory molecules and cytokines. Panel C: Shows an autoreactive B cell that has taken up autoantigens from an apoptotic cell through its antibody receptors. The B cell is receiving help from an activated T cell, which is expressing co-stimulatory molecules and cytokines involved in the maturation of B cells, including an important member of the tumor necrosis family, B lymphocyte stimulator (BLyS), also referred to as zTNF-4. The autoreactive B cell divides and matures into a plasma cell that secretes autoantibodies. It is likely that in the majority of patients, systemic lupus erythematosus develops only in the presence of abnormalities in more than one of these steps. (Adapted from Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–4.) [Reproduced with permission from Bellanti, JA (Ed). Immunology IV: Clinical Applications in Health and Disease. I Care Press, Bethesda, MD, 2012].
- Panel A shows the normal functioning of complement in facilitating the removal of an apoptotic cell without antigen presentation and inflammation.
- This is the major step which would fail to occur with an early complement component defect and/or if the system were overwhelmed.
- In panel B, as a result of complement deficiency, the apoptotic cell is not ingested normally by macrophages and instead results in maturation of a dendritic cell that then, together with the stimulatory cytokines IL-4 and IL-12, can present antigen to T cells with resultant enhanced autoreactivity.
- Other receptor-ligand systems, particularly those involving phosphoserine, may also contribute to suppression of the inflammatory/immunostimulatory consequences of phagocytosis of apoptotic cells.
- In panel C, a self-reactive B cell that has bound intracellular antigens is stimulated to differentiate into an autoantibody-secreting plasma cell.
- Since most patients with SLE do not have a complete deficiency of any individual complement component, it is presumed that partial abnormalities in regulatory mechanisms may contribute to the expression of the final disease phenotype.
- However, the occurrence of SLE in more than 90 percent of patients with C1q deficiency strongly suggests that this defect alone is sufficient to cause this multisystem autoimmune disease.
- It is remarkable that the spectrum of autoantibodies in patients with SLE is typically directed to proteins and nucleic acid antigens present in virtually every cell in the body.
- Experiments in mice suggest that in addition to complement deficiency, deficiencies in proteins that complex with or degrade chromatin are also associated with SLE-like syndromes.
Autoimmune Bullous Skin Diseases – Pemphigus
- Pemphigus is a group of organ-specific autoimmune diseases characterized clinically by the development of blisters and erosions of skin and mucosa and histologically by loss of keratinocyte adhesion also named acantholysis.
- The Pemphigus group encompasses many clinical presentations with two major forms: pemphigus foliaceus and pemphigus vulgaris characterized by sub corneal and supra basal acantholysis respectively.
- Pemphigus occurs sporadically worldwide. Endemic forms of the disease were reported in Brazil called “fogoselvagem” and in Tunisia (reaching 15 cases/106 habitants/year). This endemic form is characterized by feminine predominance at young age mainly observed in rural regions of the south of our country.
- The role of environmental factors in pemphigus is illustrated by fogoselvage, where epidemiological studies indicate that the disease is linked to exposure to environmental antigens. In Tunisia, epidemiologic studies suggested that traditional cosmetics, contact with ruminants and wasp stings are significantly associated with pemphigus.
- There are also many arguments to consider that pemphigus has a genetic basis and belong, as most organ and non-organ-specific autoimmune diseases to “polygenic” disorders whose phenotype does not result from a Mendelian inheritance pattern but from several different genes with additive or interacting effect (epistasis). The major histocompatibility complex in particular the class II locus was demonstrated to be associated with pemphigus (DRB1*0402-DQ-B1*0352 and DRB1*1401-DQ-B1*0503): see Table 1:
Table 1. Disease associations with MHC antigens
| Disease | Allele(s) associated with susceptibility | Relative Risk |
|---|---|---|
| Addison's disease | DR3 | 6 |
| Ankylosing spondylitis | B27 | 70–100 |
| Behcet's syndrome | B51 | 3–6 |
| Celiac disease | DR3, DQA1*0501, DQβ1*0201 | > 200 |
| Congenital adrenal hyperplasia | B47 | 15 |
| Dermatitis herpetiformis | DR3 | 15–18 |
| Goodpasture syndrome | DR2 | 16–20 |
| Graves' disease | DR3 | 4 |
| Hashimoto's disease | DR11 | 3 |
| Hereditary hemochromatosis | A3/B14 | 90 |
| Insulin-dependent diabetes mellitus | B35, Cw04 | 1–3 |
| Idiopathic membranous glomerulonephritis | DR3 | 12 |
| Multiple sclerosis | DR2, DQ6 | 5–12 |
| Myasthenia gravis | DR3 | 10 |
| Narcolepsy | DR2, DQβ1*0602 | 130 |
| Psoriasis vulgaris | Cw6 | 13 |
| Pemphigus vulgaris | DRβ1*0402-DQβ1*0302 | 14–21 |
| DRβ1*1401-DQβ1*0503 | ||
| Rheumatoid arthritis | DR4 | 4–10 |
| Systemic lupus erythematosus | DR3 | 3–6 |
| Sarcoidosis | DRβ1*1101 | 1.5–3.6 |
- Polymorphisms of immunoglobulins, desmogleins and cytokine genes have also been described as risk factors for pemphigus.
- Pemphigus has the prototype of an autoimmune disease, mediated by pathogenic autoantibodies directed against desmogleins (Dsg): Dsg1 in Pemphigus foliaceus and Dsg3 in Pemphigus vulgaris.
- Dsg are keratinocyte transmembrane desmosomal glycoproteins belonging to the cadherin family.
- Cadherins are keratinocyte transmembrane cell adhesion molecules that mediate Ca2+ dependent cell–cell interactions and are important for maintenance of epithelial cell integrity.
- Anti-Dsg antibodies causes acantholysis by steric hindrance, complement activation, protease activation and signal transduction.
Quiz
Related Talk
Kate Webb, Centre for Adolescent Rheumatology – Autoimmune diseases in children



