





- Patient Presentation
- History
- Differential Diagnosis
- Examination
- Investigations
- Discussion
- Treatment
- Final Outcome
- References
- Evaluation - Questions & answers
- MCQ
Patient Presentation
A severely undernourished 4-year-old girl is referred to the paediatric outpatient clinic for recurrent anaemia, an erythematous rash on her face and trunk and accompanying hepatosplenomegally.
Acknowledgment
This case study was kindly provided by Dr R Thejpal, Dr Y Goga, Dr B Neethling and Dr F Mamdoo, Specialists at Inkosi Albert Luthuli Hospital and also affiliated to the Department of Paediatrics, UKZN.
History
The child was born term, normal vaginal delivery. Although no complications were noted, her Apgar scores were recorded as 5, 6 and 8 at birth, 5 and 10 minutes respectively. Since birth the child was noted to have dysmorphic features that include most notably hypertelorism and small palpebral fissures. Her mother denies any history of antenatal alcohol use or any infections during her pregnancy. This was the first pregnancy and child born to both parents. The child was breastfed until 4 months of age; thereafter she was put on formula feeds and started on solid food by 9 months.
Her developmental milestones were delayed for both gross and fine motor skills and verbal development.
Past medical history
The child had been admitted to hospital at age 2½ years for lower respiratory tract infection (LRTI), suspected of being TB. The child was also failing to thrive, falling below the 5th centile for both height and weight. At this time she was investigated for TB, Epstein- Barr Virus (EBV), Toxoplasmosis, Rubella, Cytomegalovirus (CMV), Herpes, Hepatitis B and HIV. She was found to be negative for all investigations except EBV, which was IgM negative but IgG positive. She was further tested for Ceruloplasmin 0.69 (0.18-0.45) and copper levels 35.9mmol/l, which were both mildly elevated. Her complement C3 was 2.2 (0.8-1.5) and C4 was 0.90 (0.84), which were both elevated.
A year later at age 3½ years she was re-admitted for LRTI, gastric washings were submitted but found to be TB negative, while complement C3 and C4 levels remained elevated at the same levels recorded a year earlier.
Past surgical history
Nil noted
Family history
No positive TB contacts. Both parents are HIV negative and in good health.
No siblings or other children in the home
Travel history
None
Social history
Father is employed as a security guard and mother is currently unemployed. They live together in a one bedroom apartment with electricity and running water. No pets and no smoking in the home
Medication
Nil
Allergies
Nil known
Differential Diagnosis
- HIV
- TB
- Recurrent pneumonia
- Cystic fibrosis
- Hypogammaglobulinaemia
- Agammaglobulinaemia
- Severe combined immunodeficiency (SCID)
Examination
General
- Weight 8.3 kg, below 5th centile
- Height 75cm, below 5th centile
- Afebrile
- Blood pressure and heart rate normal
- Moderate pallor
- Generalised lymphadenopathy with some firm axillary lymph nodes bilaterally
- No dehydration
- No jaundice
- No stigmata of HIV
Head and Neck
- Dysmorphic features- hypertelorism and small palpebral fissures
- Puffy eyelids
- Anterior and posterior fontanelles open
- Hair has red discolouration and straight
- Erythematous rash with mild induration noted on parts of the face, forehead and trunk- thought to be atypical erythema nodosum
Abdomen
- Distended, with some scratch marks noted
- Soft and non tender on palpation
- Large hepatosplenomegaly
- Bowel sounds heard on auscultation
Respiratory
- No signs of respiratory distress
- Chest clear
Cardiovascular
- Ejection systolic murmur but no significant cardiac lesions
Neurological
- Delayed milestones with regards to motor skills (fine and gross) and verbal skills. Higher function could not be assessed because child did not communicate adequately
- Reflexes 2/4
- Sensation intact
- Tone normal
- Strength normal
- Gait normal
Musculoskeletal
- Areas of induration under the skin of both calves, possibly due to calcification in the calf muscles.
Investigations
| Admission Age 3 ½ years | Current Admission | |||
|---|---|---|---|---|
| Age 4 years | Day 5 | Day 8 | ||
| Total protein | 67 g/l | 62 g/l | ||
| Albumin | 37 g/l | 32 g/l | ||
| Total Bilirubin | 12.0 umol/l | 15.0 umol/l | ||
| Direct Bilirubin | 2.0 umol/l | 2.0 umol/l | ||
| Alk phos. | 299 U/l | 194 U/l | ||
| Gamma GT | 213 U/l | 77 U/l | ||
| ALT | 43 U/l | 45 U/l | ||
| AST | 88 U/l | 80 U/l | ||
| IgG | 12.90g/l | |||
| IgA | 1.30 g/l | |||
| IgM | 2.30 g/l | |||
| Ceruloplasmin | 1.0900000000000001 | |||
| ANF neg | ||||
| WCC | 2.8 | 4.2 | 2.5 | |
| Hb | 5.4 | 5.9 | 6.4 | |
| MCV | 84 | |||
| Plt | 80 | 52 | 43 | 20 |
| Differential | ||||
| Neutrophils | 1.44 | |||
| Lymphocytes | 0.25 | |||
| Monocytes | 0.37 | |||
| Band% | 0.42 | |||
| Band absolute | 1.5 | |||
| Myelo % | 0.01 | |||
| Myelo absolute | 0 | |||
| Smudge cells % | 0.01 | |||
| Smudge cells absolute | 0.03 | |||
| NRBC | 1.6 | |||
| HIV | Negative | Negative | ||
| Parvovirus PCR from bone marrow aspirate | Positive | |||
| Urine metabolic screen | negative | |||
| Immune profile | ||||
| CD3% | 0.33 | |||
| CD3 | 85 cells/ul | |||
| CD4% | 0.13800000000000001 | |||
| CD4 | 35 cells/ul | |||
| CD8% | 0.251 | |||
| CD8 | 65 cells | |||
| CD19% | 1.4E-2 | |||
| CD19 | 3 cells/ul | |||
| CD56% | 0.53600000000000003 | |||
| CD56 | 138 cells/ul | |||
| CD10% | 1.6999999999999999E-3 |
Interpretation: T and B cell lymphopaenia noted
Preservation of natural killer cells
Suggestive of a primary immune deficiency
Current Admission
Bone marrow:
The aspirate failed, but the trephine was suggestive of mycobacterial infection with secondary marrow fibrosis. In addition, one of the TB cultures were Zn positive, however the final culture yielded no growth (could be MOTTS or MTB).
Liver biopsy:
Mild portal tract inflammation with eosinophils, extramedullary haemopoeisis and focal lobular inflammation; Mild macrovesicular steatosis; Liver biopsy result is in keeping with mild portal tract chronic inflammation with one non-necrotising granuloma on deeper sections, Ziehl Nielsen stains for AFB negative.
Skin Biopsy:
Skin with deep seated histiocytic infiltrate within the subcutaneous tissue. CD1a and S100 are negative excluding a Langerhans cell infiltrate; Chloroacetate esterase stain is positive in some of the immature myeloid cells. Stains for AFB and fungi are negative.
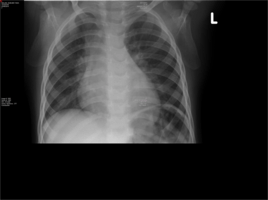
Chest X-ray:
Pneumonitis of both lung fields
Discussion
This case explores the clinical manifestation of primary immunodeficiency, specifically examining a type of severe combined immunodeficiency (SCID). SCID is a life-threatening syndrome of recurrent infections, diarrhoea, skin infections and failure to thrive that results from numerous molecular defects leading to severe T- and B-cell dysfunction and occasionally can affect natural killer (NK) cells. Without intervention, the T and B-cell dysfunction usually results in severe infection and death in children by age 2 years.
Suspected SCID is defined as less than 0.3×109/L CD3 T cells or less than 20% of CD3/CD4 cells with naïve cell markers with one or more of the following: (1) abnormal TRECs (T-cell receptor excision circle) upon new born screening or presentation, (2) family history of SCID and (3) recurrent or opportunistic infections. Features of Omenn syndrome is also an early indicator (Severe Combined Immunodeficiency (SCID) | NIH: National Institute of Allergy and Infectious Diseases, 2015.). Clinically, most patients present before age 3 months with unusually severe and frequent infections by common or opportunistic pathogens. This is obviously not the situation with our patient, but we will explore the mechanism and pathophysiology of SCID so that a likely scenario can be proposed with the 4-year-old girl in our case. It is first important to understand T and B-cells and the way in which they generate immunological diversity in a normally functioning immune system. All mechanisms described here are carefully illustrated in stepwise graphics to assist with understanding the processes described.
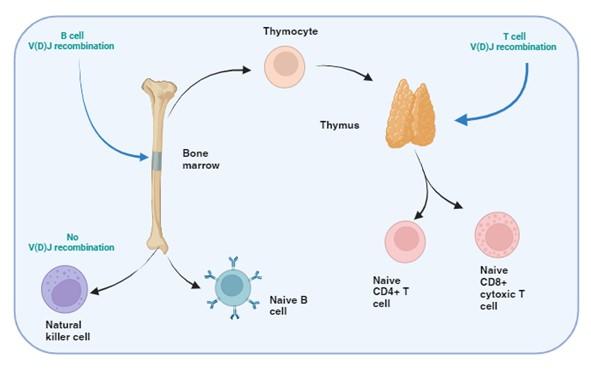
Figure 1. T and B cell V(D)J recombination. The image illustrates the specific tissue in which B and T cell recombination occurs, allowing BCR and TCR to recognize a wide range of pathogenic epitopes. Variability in epitope binding potential of the receptors is achieved by varying the combination of pre-existing multicopy gene segments called variable (V), diversity (D) and joining (J) segments found in the BCR-heavy and TCR-β and -δ chains. In B cells the process take place in the bone marrow and for T cells in the thymus. Created with biorender.com (CA Petersen 2024)
How is diversity normally generated in T and B cells?
T and B cells, but not NK cells, undergo V(D)J recombination in order to generate diverse repertoires of T and B cell receptors (TCR and BCR) capable of recognising a wide range of pathogens. This is achieved by each TCR and BCR being highly specific for small protein fragments derived from the pathogen. These small protein fragments are known as epitopes. As there are potentially an infinite number of epitopes, the TCR and BCR need to be adaptable and variable so that enough diversity is created to engage with each different epitope.
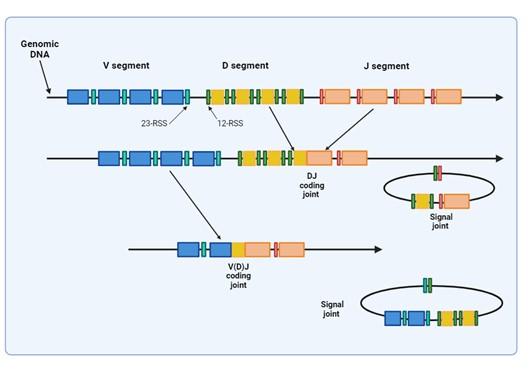
Figure 2. Illustrates how VDJ recombination occurs in the BCR-H, TCR-β and -δ chains. The BCR-L, TCR-α and -γ chains lack D segments and only undergo VJ recombination. Recombination activating genes (RAG-1 and RAG-2) mediates the recognition and splicing of the V, D and J segments. Created with biorender.com (CA Petersen 2024).
Variability in the epitope binding potential of the receptors is achieved by varying the combination of pre-existing genes known as Variable (V), Diversity (D) and Joining (J) segments. BCR (B cells) variability takes place in the bone marrow and TCR (T cells) variability occurs in the thymus. Variability and generation of diversity occurs by V(D)J gene recombination in the different anatomical locations. Expression of these recombined genes in the B cell makes up the protein structure of the BCRheavy chain and in the T cell makes up the TCR-b and -d chains. Diversity (D) segments are absent in the BCR-light (k and l) and in the TCR-a and -g chains, so only V and J recombination occurs for these proteins.
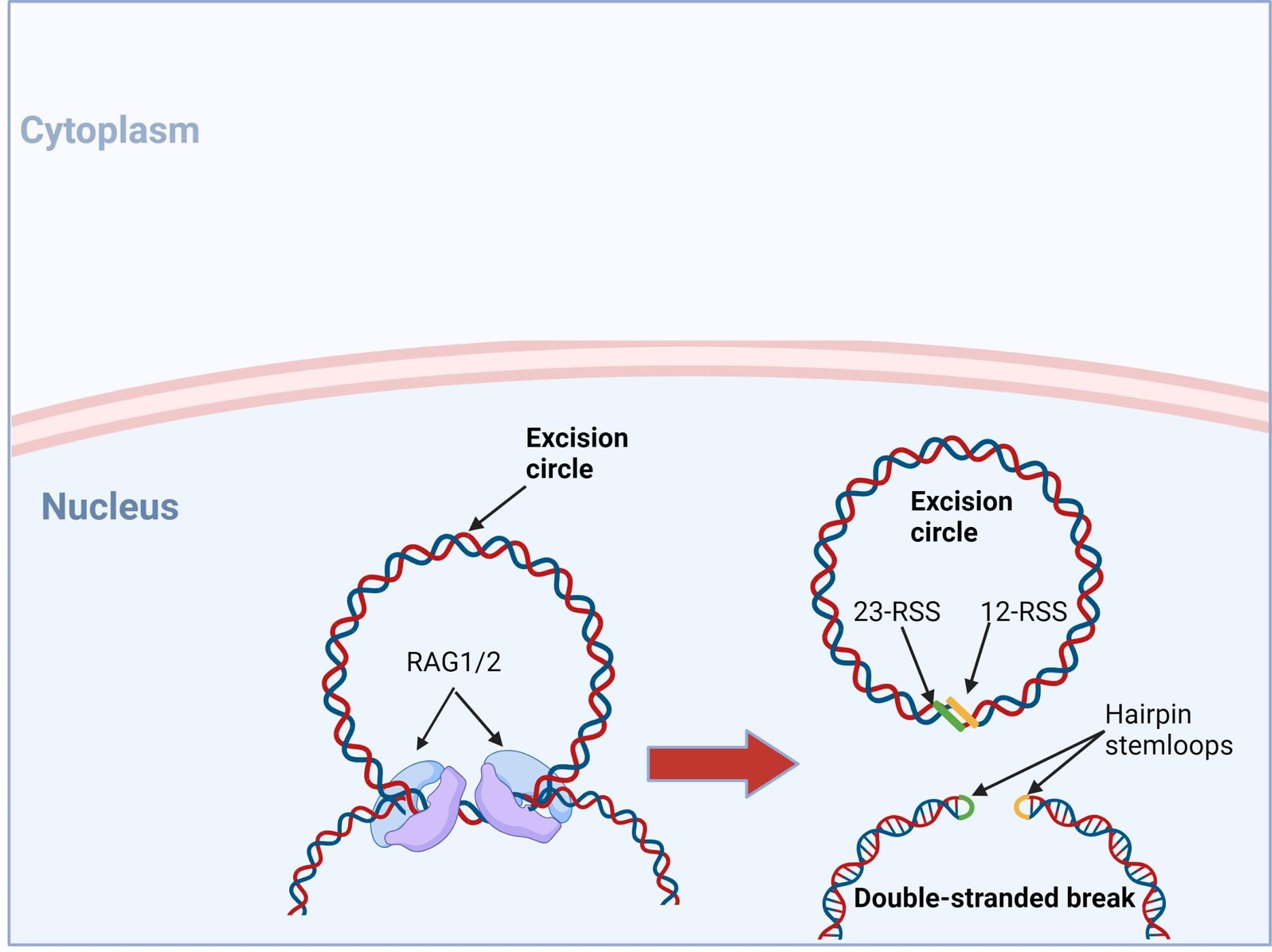
Figure 3. The RAG1/2 complex mediates the random selection and alignment of a D and J or V and DJ gene segment and introducing a double strand break in the DNA. The blunt-ends of the excision circle are ligated between the 12-RSS and 23-RSS sequences to generate the signal joint, while the 5’ and 3’ ends of the genomic DNA strands are covalently linked to generate hairpin stem loop structures. Created with biorender.com (CA Petersen 2024).
Let’s look a little closer at V(D)J recombination
VDJ recombination occurs in the BCR-H, the TCR-β and -δ chains, whilst only VJ recombination occurs in the BCR-L (k and l) and TCR-a and -g chains. Gene recombination can occur because of Recombination Activating Genes that encode two enzymes: RAG1 and RAG2. These mediate the binding and splicing of V, D and J segments. RAG1 binds to specific recombination signal sequences (RSS) present as 23-RSS or 12RSS sequences, which flank each gene segment, and then recruit RAG2. Recombination can only occur between a 23-RSS and 12-RSS sequence (which is called the 12/23 rule). A double-stranded break is introduced into DNA followed by recruitment of DNA repair enzymes that mediate Non-Homologous End-Joining (NHEJ). Recognition and end-binding is mediated by proteins Ku70, Ku80 which recruit DNA-PKcs.
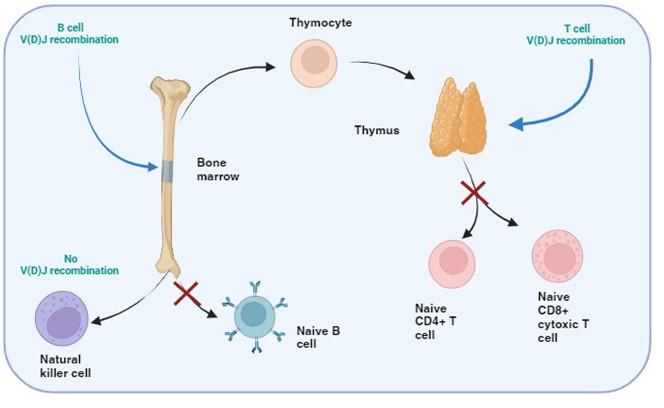
Figure 4. SCID: defective T and B cell development, normal NK cells. In certain types of SCID, defect in the development of T and B cells, but normal NK cells function is evident. This stems from defects in the V(D)J recombination processes common to T and b cell maturation, but is not required for NK cell development. Created with biorender.com (CA Petersen 2024).
There are many proteins involved in V(D)J recombination and to date genetic mutations in RAG1, RAG2, DNA-PKcs, DCLRE1C (also known as Artemis), XLF (also known as Cernunnos) and DNA ligase IV have been identified in the SCID phenotype that result in a lack of T and B cells, but with normal NK cell activity. The most common mutation is seen in RAG1 and RAG2.
According to the Primary Immune Deficiency Treatment consortium (PIDTC) 2022 definition for the diagnosis of SCID, there are three major subtypes: typical SCID with very low T cells, leaky SCID with low T cells and Omenn syndrome. Typical SCID describes patients with the most profound defect in host T cells, usually due to null pathogenic variants in genes whose products are essential for T cell development. Defects in 7 known genes (IL2RG, RAG1, RAG2, ADA, DCLRE1C IL7R and JAK3) represents 80% of SCID cases. A pathogenic finding in many genotypes of typical SCID is the presence of maternal T cells in the peripheral blood, this is due to the failure to reject transplacental transferred cells. Transplacental acquired maternal engraftment (TME) is approximately observed in 50% of patients with typical SCID but somewhat less in patients with a genetic subtype ADA, RAG1, RAG2 and DCLRE1C, which is likely due to the ability of NK cells and residual host T cells to eliminate the maternal cells. TME may elevate the total amount of T cells, so in the absence of TME the threshold for typical SCID diagnosis would less than 0.05 x109/L CD3 T cells according to the PIDTC 2022 (Dvorak et al., 2023).
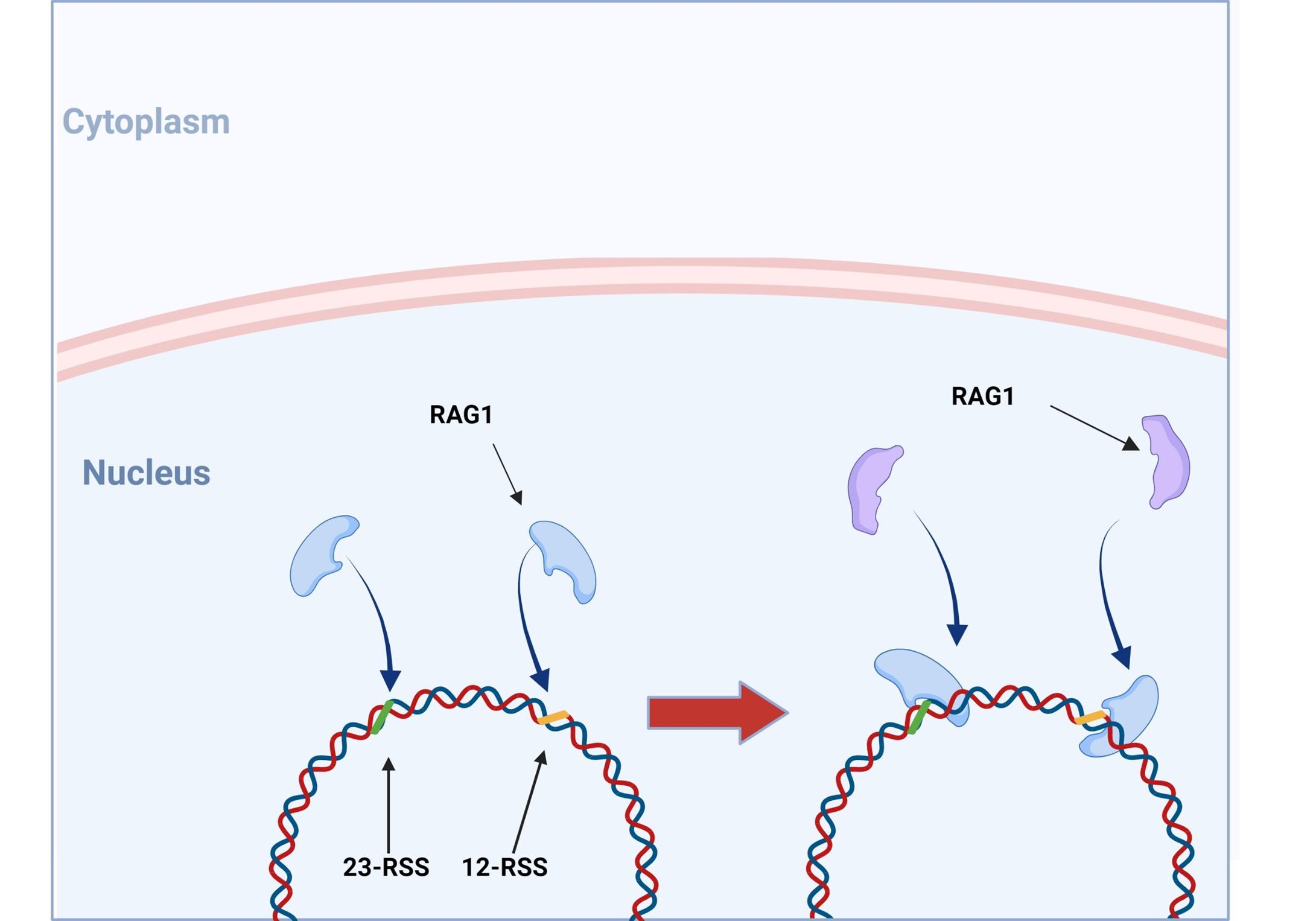
Figure 5. V(D)J recombination: RAG1 and RAG2 binding. V(DJ recombination begins with the binding of recombination activating gene 1 (RAG1) proteins to specific recombination signals sequences (RSS) present in each V, D and J segment. RAG2 is then recruited and the RAG1/2 complex mediates the formation of a double stranded beak in DNA which will allow a precise fusion if a D and J or V and (D)J segment. Created with biorender.com (CA Petersen 2024)
Worth noting is that in certain rare forms of SCID, with defects in T and B cell development, the genetic mutations in the affected proteins may permit partial enzymatic activity, which is then known as “leaky” SCID. Defects in RAG1, RAG2, ADA and RMRP are overrepresented in leaky SCID. Leaky SCID diagnosis requires at least 2 of the following: low T cell number s for age (<0.6×109/L for any age, <0.8×109/L if aged 2-4 years or <1.0×109/L if aged < 2 years), an oligoclonal T cell population and low percentage of naive T cells or low/undetectable TRECs. It is also critical to test for TME, because maternal cells would classify the patient as typical SCID (Dvorak et al., 2023)
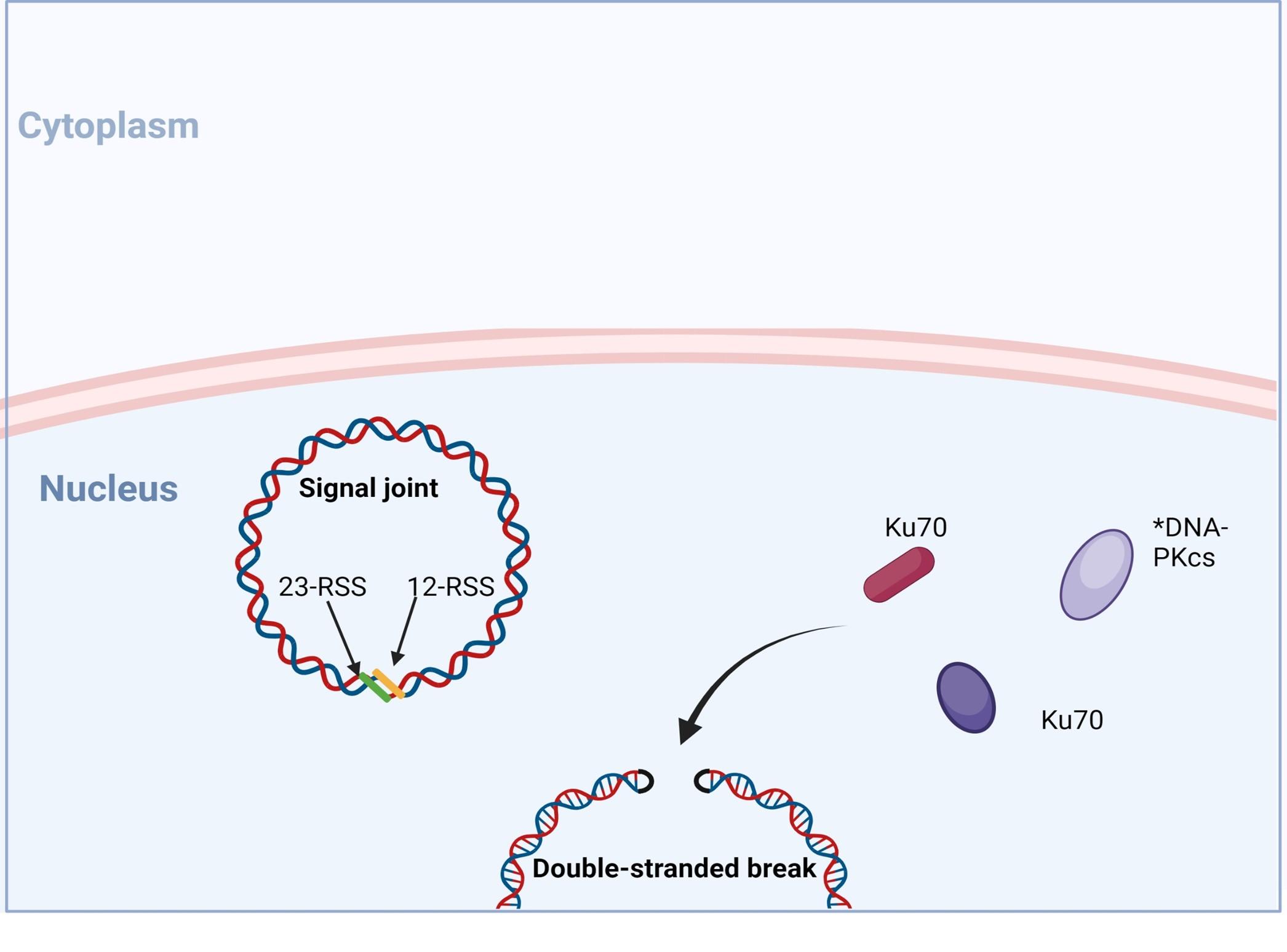
Figure 6. No- homologous end joining: recognition and end binding. Following the introduction of double stranded break in the genomic DNA RAG1/2 complex, recruitment of DNA repair enzymes is initiated to mediate non-homologous end joining (NHEJ) of DNA ends. Recognition and end-binding is first mediated by proteins Ku70, Ku80 which recruit DNA-PKs. *In SCID phenotypes with defective T and B cell development, but normal NK cell function, genetic mutation in DNA-PKcs have been identified Created with biorender.com (CA Petersen 2024)
It has been reported in Omenn’s syndrome that clonal expansion of activated T and B cells occurs and that blood levels of these cells may appear within normal ranges, or be elevated. Closer examination the TCR and BCR in these patients reveal them to be oligoclonal in nature. Oligoclonal refers to there only being very few BCR or TCR types because there is an inability to undergo complete V(D)J recombination, and the result is translated to a limited recognition of epitopes. This is extremely dangerous for the patient as this indicates that there is not enough BCR or TCR diversity to interact with the array of epitopes required to mount an effective immune response, but also because the epitopes recognized tend to be directed to self (autoimmunity). The resulting clinical profile is immunodeficiency along with autoimmunity. This produces an erythematous rash, often associated with lymphadenopathy, hepatosplenomegaly and other clinical features. Exclusion of TME is crucial for making a diagnosis of Omenn syndrome, as most patients are initially diagnosed with typical SCID, that later develops into Omenn syndrome, thus patients should be monitored for development of Omenn syndrome over time.
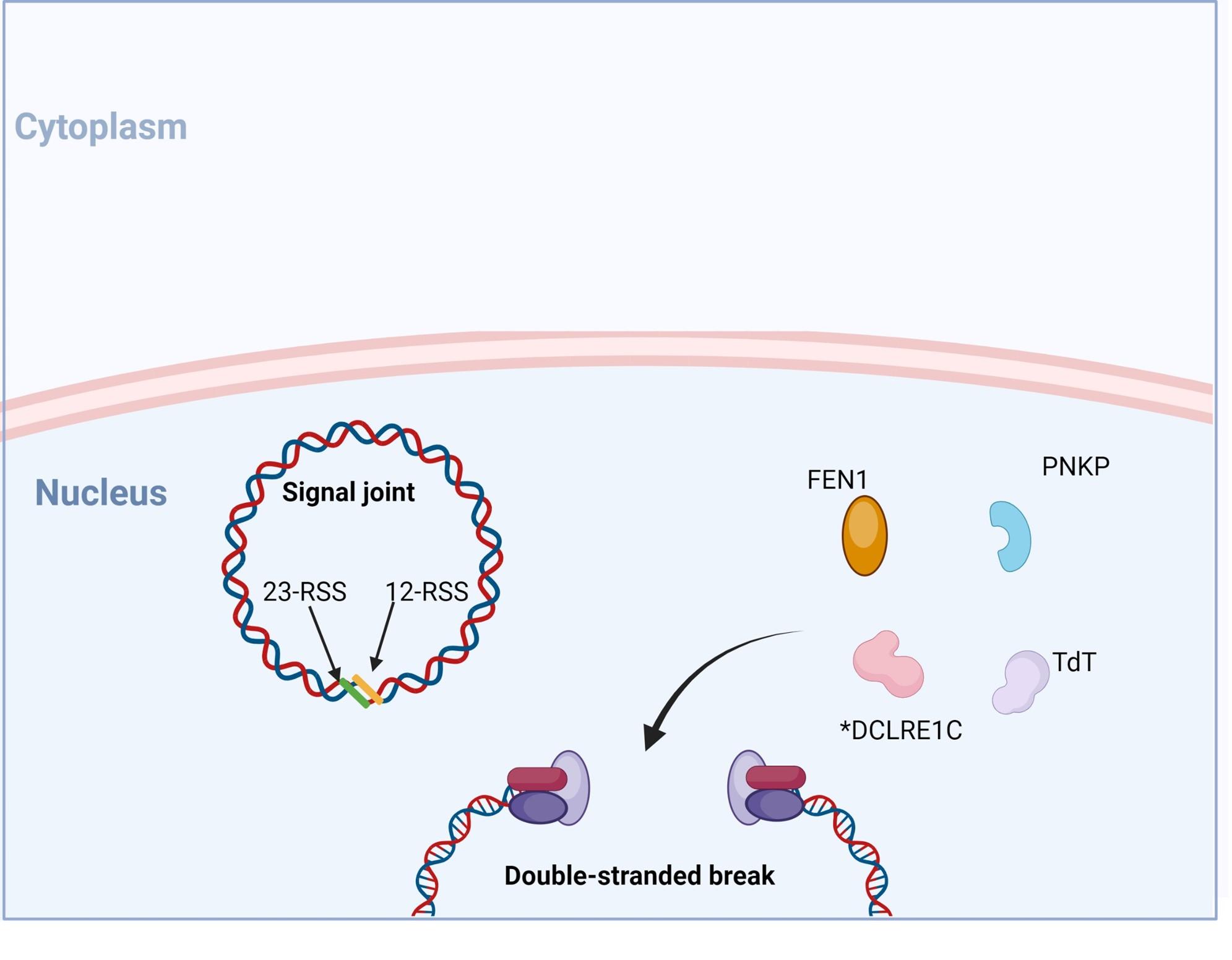
Figure 7. Following recognition and binding of the DNA ends, recruitment of proteins FEN, DCLRE1C (also known as Artemis), Tdt and PNKP occurs. DCLRE1C mediates nicking of the hairpin structures at random nucleotides while TdT adds random nucleotides to the 3’ end. This process provides an additional mechanism of introducing variability into the coding regions of the TCR and BCR known as junctional diversity. Created with biorender.com (CA Petersen 2024)
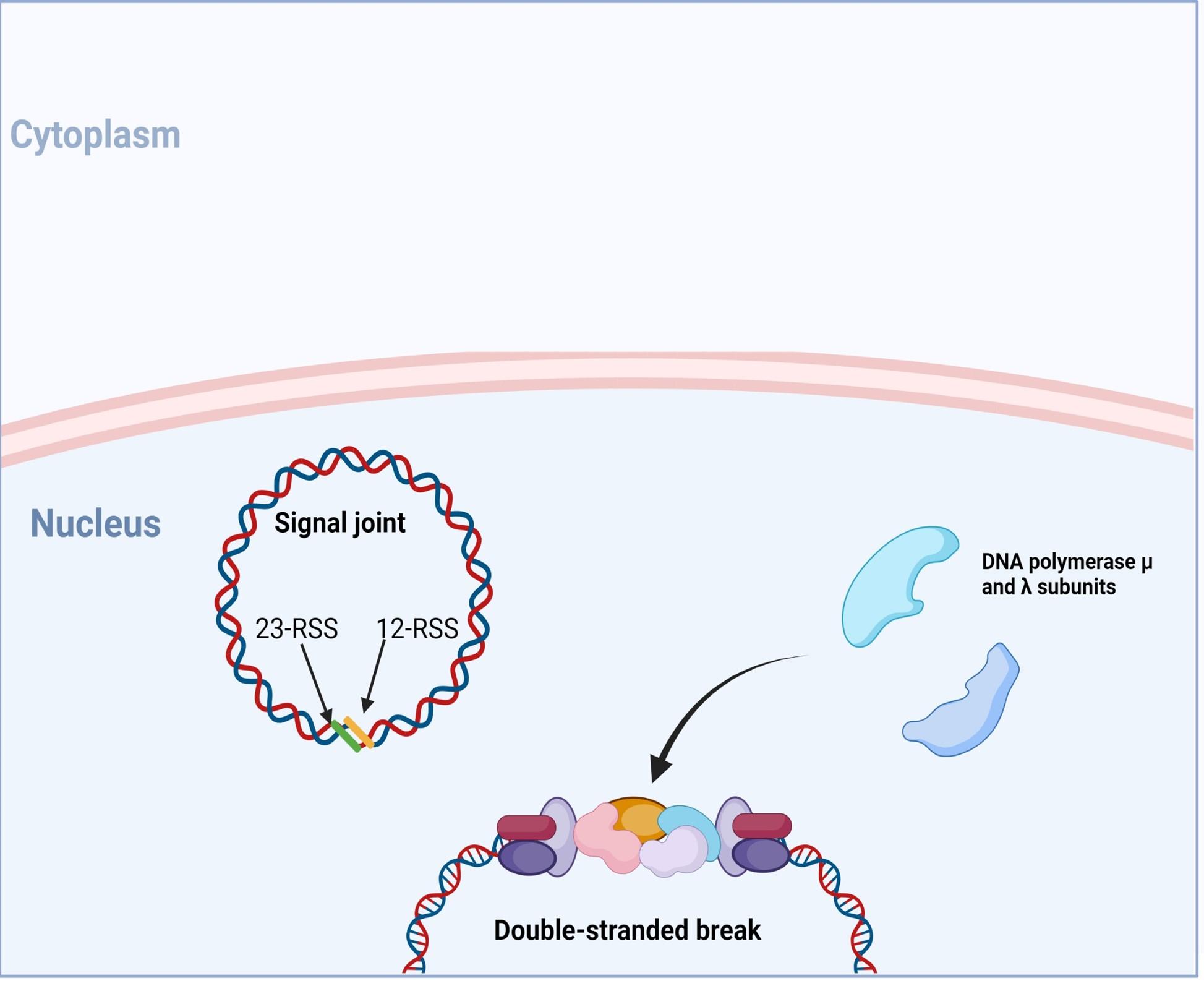
Figure 8. Following end-processing, DNA polymerase µ and ʎ are recruited to fill in single -stranded gaps in the DNA strands. Created with biorender.com (CA Petersen 2024)
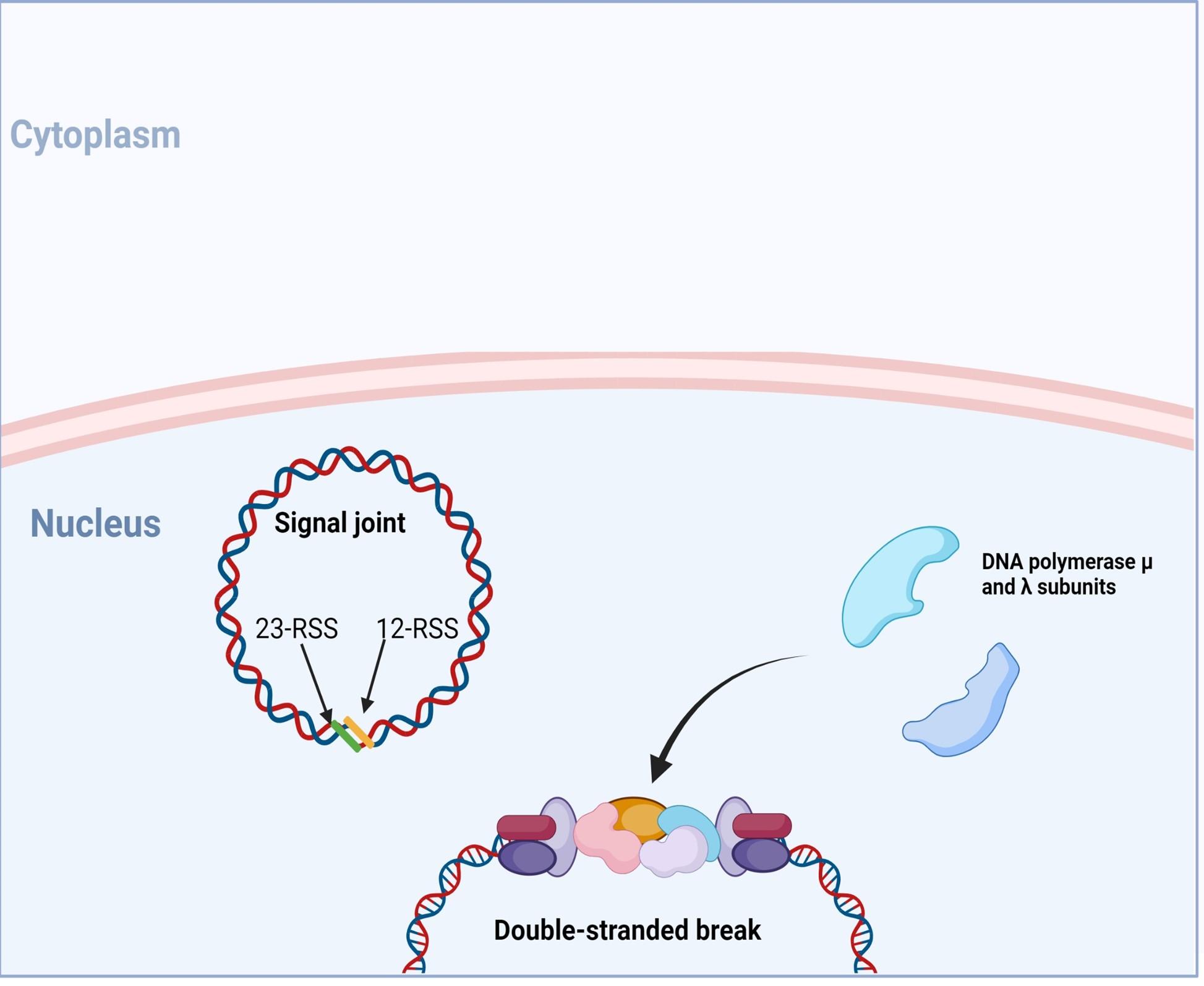
Figure 9. following DNA polymerase, a final ligation of the 3’-OH and 5’-P ends is initiated and requires recruitment of proteins XRCC4, XLF (also known as Cernunnos) and DNA ligase IV. *In SCID phenotypes with defects in T and B cells development, but normal NK cell function, genetic mutations in XLF and DNA ligase IV have been identified. Created with biorender.com (CA Petersen 2024)
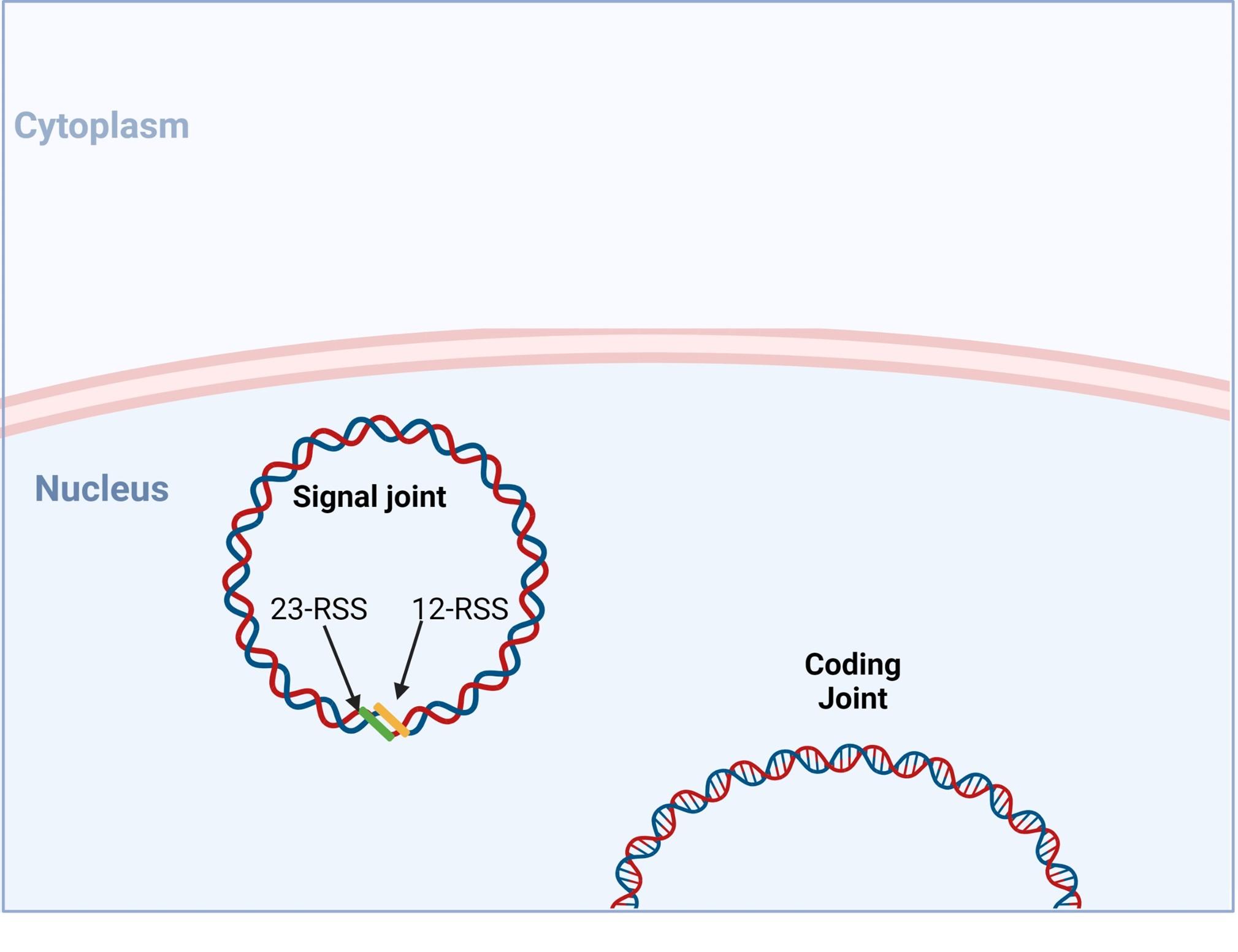
Figure 10. V(D) recombination is completed when a functional coding joint has be generated. Created with biorender.com (CA Petersen 2024)
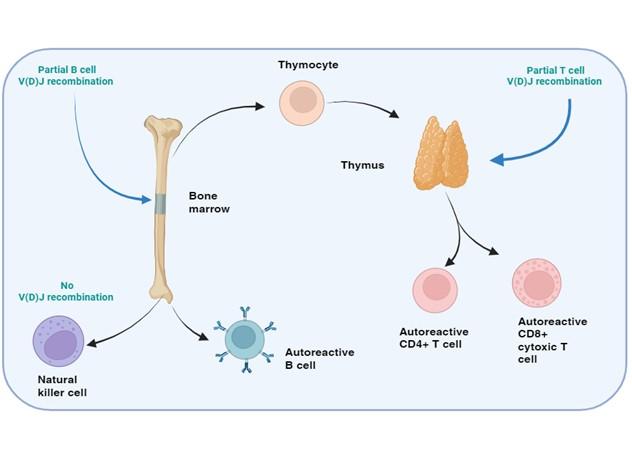
Figure 11. SCID with autoimmunity: Omenn’s syndrome. In certain rare forms of SCID with defects in the T and B cell development, but normal NK cell function, the genetic mutations in the affected proteins may permit partial enzymatic activity, known as “leaky” SCID. The restrictive V(D)J recombination permits the development of a limited repertoire of T and B cells which may be directed towards self-antigens leading to an autoimmune response, know as Omenn’s syndrome. Clonal expansion of theses activated T and B cells occurs and blood levels of these cells may appear in range or elevated. Phenotypic examination of the TCR or BCR, however reveals that these cells are oligoclonal in nature. Created with biorender.com (CA Petersen 2024)
Treatment
Current case
The patient was transfused with packed red cells to assist with the severe anaemia. IV immunoglobulin prophylaxis (polygamy) was given during admission. Empiric antibiotic therapy was started at admission as well as four-drug TB treatment initiated.
Bone marrow transplantation
The only cure for SCID is bone marrow transplantation. Extreme care to match the HLA of the donor with the host is the optimal option (usually a living related donor), but an HLA-matched unrelated donor and even HLA partial-mismatched related donor transplantation can be considered with successful outcomes. This approach is successful if the disease is diagnosed within their first 3 months of life and early transplantation before 3.5 months is associated with better overall survival.
Vaccinations
No live vaccines, such as the BCG vaccine, should be administered to patients with SCID prior to bone marrow transplantation.
Intravenous immunoglobulin
The therapeutic function is passive immunization to prevent infection. IVIG can be used to restore antibody levels until the B-cell system is restored with transplantation. However, long-term use fails to change the terminal course of SCID.
With the advances in bone marrow transplantation and gene therapy, patients now have a better likelihood of developing a functional immune system in a previously lethal genetic disease. However, once an infant develops serious infections, intervention is rarely successful
Final Outcome
Unknown. After discharge patient failed to return for follow up and no further hospital admissions have been noted.
References
- Niehues T et al. (2010). More than just SCID–the phenotypic range of combined immunodeficiencies associated with mutations in the recombinase activating genes (RAG) 1 and 2. Clin Immunol. 2010 May;135(2):183-92. Epub 2010 Feb 20
- Gennery AR et al. (2005). Omenn’s syndrome occurring in patients without mutations in recombination activating genes. Clin Immunol. 2005 Sep;116(3):246-56.
- Notarongelo LD. (2010). Primary immunodeficiencies. J Allergy Clin Immunol. Feb 2010;125 (2 suppl):S182-94
- Dvorak, C. C., Haddad, E., Heimall, J., Dunn, E., Buckley, R. H., Kohn, D. B., Cowan, M. J., Pai, S. Y., Griffith, L. M., Cuvelier, G. D. E., Eissa, H., Shah, A. J., O’Reilly, R. J., Pulsipher, M. A., Wright, N. A. M., Abraham, R. S., Satter, L. F., Notarangelo, L. D., & Puck, J. M. (2023). The diagnosis of severe combined immunodeficiency (SCID): The Primary Immune Deficiency Treatment Consortium (PIDTC) 2022 Definitions. Journal of Allergy and Clinical Immunology, 151(2), 539–546. https://doi.org/10.1016/j.jaci.2022.10.022
- Severe Combined Immunodeficiency (SCID) | NIH: National Institute of Allergy and Infectious Diseases. (n.d.). Retrieved October 26, 2023, from https://www.niaid.nih.gov/diseases-conditions/severe-combined-immunodeficiency-scid
- Slatter, M. A., & Gennery, A. R. (2022). Advances in the treatment of severe combined immunodeficiency. Clinical Immunology, 242, 109084. https://doi.org/10.1016/j.clim.2022.109084
Evaluation – Questions & answers
What is the diagnosis?
What is SCID?
SCID is a life-threatening syndrome of recurrent infections, diarrhoea, skin infections and failure to thrive, that results from numerous molecular defects affecting the immune system which leads to severe T- and B-cell dysfunction and occasionally affects natural killer (NK) cells. Clinically, most patients present before age 3 months with unusually severe and frequent infections by common or opportunistic pathogens. SCID is rare and occurs in one in 50,000 births. Without intervention, the T- and B-cell dysfunction usually results in severe infection and death in children by age 2 years. There are a number of different causes of SCID, with each type caused by a different genetic defect along a different pathway.
What is the typical history of a child presenting with SCID?
Often these patients will present with failure to thrive and multiple severe or recurrent illnesses with both viral and bacterial causes such as otitis media, diarrhoea and dermatitis during the first 3 months of life. They also present commonly with pneumonia and mucocutaneous candidiasis which is severe and resistant to treatment. The viral infections typically seen in these patients include varicella, herpes simplex, respiratory syncytial virus (RSV), rotavirus, adenovirus, enterovirus, parainfluenza virus, Epstein-Barr virus (EBV), and cytomegalovirus (CMV).
What are the findings on examination of these patients?
Findings are specific for the various infections associated with patients who have SCID. Sparse hair, abnormal dentition, osteopetrosis and common cutaneous findings such as eczematous dermatitis, recurrent furunculosis, oral thrush, and herpetic dermatitis may be noted. Important to note is that patients with SCID fail to manifest palpable lymphadenopathy and this lack of recognizable peripheral lymph nodes should raise suspicion of SCID in children with multiple aggressive infections.
What is the pathophysiology of SCID?
Severe combined immunodeficiency results from mutations in one of more than 15 known genes. These molecular defects block the differentiation and proliferation of T-cells, B-cells and occasionally NK cells. Antibody production is severely impaired, even when mature B-cells are present due to lack of T-cell help. NK cells, a component of innate immunity, are variably affected. Classification of the aetiologies of SCID is according to the corresponding phenotypic lymphocyte profiles which are T-lymphocyte negative (T), B-lymphocyte
positive (B+), and NK-negative (T– B+ NK–); T– B– NK–; T– B– NK+; and T– B+ NK+.
How does SCID most commonly occur?
A wide repertoire of T-cell receptor or immunoglobulin diversity is achieved by somatic recombination of variable (V), diversity (D) and joining (J) gene segments. The recombination activating gene (RAG)-1 and RAG-2 endonuclease proteins initiate this recombination by cleaving DNA at recombination signal sequence sites. Cleaved DNA subsequently repaired by the non-lymphoid-specific DNA double- strand break repair machinery. Mutations in RAG-1 or RAG-2 genes result in a functional impairment of antigen receptor recombination, which causes T-negative B-negative severe combined immunodeficiency (T-B-SCID). This is the most commonly affected pathway resulting in SCID. However, genetic mutations can occur in other proteins involved in the recombination process which include DNA-PKcs, DCLRE1C (also known as Artemis), XLF (also known as Cernunnos) and DNA ligase IV.
Does SCID always result in a complete primary immunodeficiency where both B and T cells are completely non-functional?
Although SCID typically results in complete deficiency, rare forms do occur where the genetic mutations in the affected proteins involved in T and B- cell recombination may permit partial enzymatic activity, which is known as “leaky” SCID. This restrictive V(D)J recombination permits the development of a limited repertoire of T and B-cells which may often be directed towards self-antigens leading to an autoimmune response, known as Omenn’s syndrome. On closer examination of the TCR or BCR, it reveals that these cells are oligoclonal in nature, meaning that clonal expansion of these activated T and
B cells has occurred and blood levels of these cells may appear in range or elevated. However, because of the limited recognition of epitopes, there is still a resulting immunodeficiency.
How is new born screening used to diagnose SCID?
SCID is better managed if diagnosed early, an effective way to identify if a patient is at risk of SCID is through new born screening. During T cell development redundant gene are cleaved and stored as episomal DNA in thymocytes that does not replicate during cell division. The episomal DNA or T lymphocyte excision circle (TREC), functions as a marker of naïve T cell development. Infants with SCID have no TRECs and this marker can be detected using PCR on the new born blood spot taken five days after birth, allowing it to be used as a mass screening to test for SCID. Early detection of SCID while the infants are infection free significantly increase the success of HSCT treatment(Slatter & Gennery, 2022).
Multiple Choice Questions
Earn 1 HPCSA or 0.25 SACNASP CPD Points – Online Quiz



