





- Patient Presentation
- History
- Differential Diagnosis for Declining CD4 Count
- Examination and Investigation
- Discussion
- Final Outcome
- References
- Evaluation - Questions & answers
Patient Presentation
A 22 year old female, who was diagnosed as HIV positive 18 months ago and has been on antiretroviral treatment since diagnosis. Although she had been doing well she recently started complaining of nausea and abdominal pains.
Acknowledgement
This case study was kindly provided by Dr Harry Moultrie, Director of Enhancing Children’s HIV Outcomes (ECHO), University of the Witwatersrand and Dr Monica Mercer from Immunopaedia
History
20 years 6 months of age
The young lady presented to her local clinic following the death of her partner from AIDS. Although she appeared well and had not complained of any serious illnesses or had any prior hospital admissions she requested that she be tested for HIV.
- She was examined and found to be thin with a generalised lymphadenopathy, but was otherwise well.
- An HIV ELISA test was performed and was found to be reactive.
- She was classified as having WHO stage 1 disease
- Her CD4 count was 185 cells/ul
- She was initiated on a HAART regimen of Didanosine (DDI), Tenofovir (TDF) and efavirenz (EFV). Although this is not a typical initial regimen it was prescribed based on availability of medication.
- She was also started on cotrimoxazole prophylaxis and multivitamins
4 months later
After 4 months of antiretroviral therapy the patient had gained weight and had achieved viral suppression with her viral load measuring less than 25 copies.
- CD4 count increased to 256 cells/ul
8 months after starting treatment
She continued to do well, remained virally suppressed and her CD4 count continued to increase.
- CD4 count 311 cells/ul
10 months after starting treatment
She continued to do well, remaining virally suppressed but her CD4 count had decreased since her last test.
- CD4 count 270 cells/ul
14 months after starting treatment
She continues to do well, remaining virally suppressed but her CD4 count has continued to decrease.
- CD4 count 215 cells/ul
18 months after starting treatment
On this visit she was noted to be complaining of nausea and abdominal pain radiating to the back. Her viral load was tested and was found to be undetectable, but her CD4 count had continued to decrease dropping below baseline.
- CD4 count 165 cells/ul
Her lipase and amylase levels were also tested and found to be elevated, confirming the clinical diagnosis of acute pancreatitis.
The pancreatitis was most likely due to Didanosine (DDI) toxicity. Her drug regimen was therefore changed to Tenofovir (TDF), Zidovudine (AZT) and efavefairenz (EFV)
22 months after starting treatment
The acute pancreatitis resolved, she remained virally suppressed and her CD4 count had once again increased above her baseline.
- CD4 count 280 cells/ul
26 months after starting treatment
She continues to do well, remains virally suppressed and her CD4 count has increased.
- CD4 count 322 cells/ul
Differential Diagnosis for Declining CD4 Count
- Laboratory error
- Drug resistant replicating virus in lymph nodes, but undetectable in plasma
- Non compliance
- Drug toxicity
| Months on HAART | Start | 4 | 8 | 10 | 14 | 18 | Change of Regimen | 22 | 26 |
|---|---|---|---|---|---|---|---|---|---|
| CD4 | 185 | 256 | 311 | 270 | 215 | 165 | 280 | 322 | |
| Viral Load | 231000 | ||||||||
| TB status-sputum culture | Neg | Neg | Neg | Neg | |||||
| MDR-TB | Neg | Neg | |||||||
| WHO clinical stage | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Side effects experienced | Nausea and abdominal pain - pancreatitis | ||||||||
| Opportunistic Infections | Nil | Nil | Nil | Nil | Nil | Nil | Nil | Nil |
Discussion
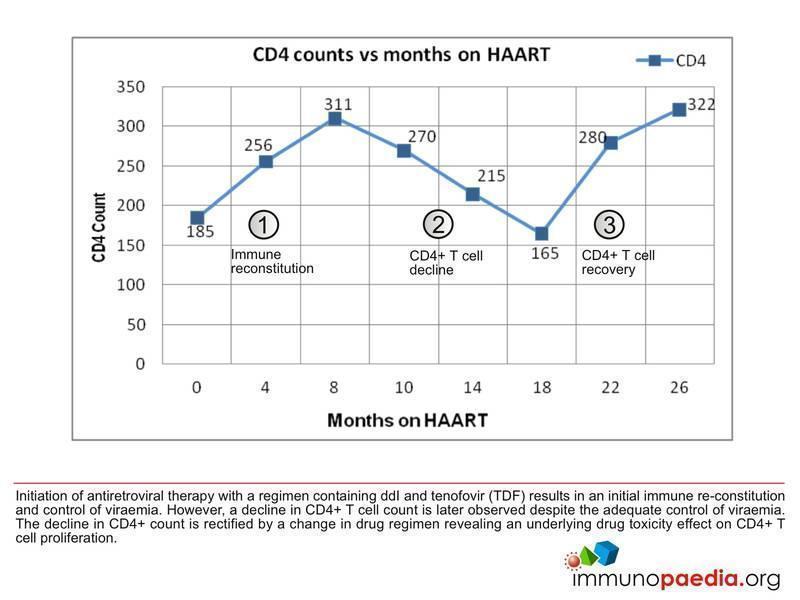
There are three immunologic phases which can be described as occurring in this patient. Firstly, there is the initiation of antiretroviral therapy with a regimen containing Didanosine (ddI) and tenofovir (TDF), which results in immune reconstitution and control of viraemia. However, this is followed by a decline in the absolute CD4 T cell count despite the adequate control of viral load. The decline in CD4 counts is then rectified by a change in drug regimen, which reveals an underlying drug toxicity effect on the CD4+ T cell population.
Each of these processes is discussed in more detail:
1) Immune reconstitution
Immune reconstitution is the initial increase in peripheral blood absolute CD4+ T counts following the initiation of antiretroviral therapy. This is observed to occur during the first 6 months of treatment with memory CD4+ T-cells taken to be be the highest contributors. This can be considered a restoration process, where the immune system is re-established due to suppression of replicating HIV by the effective drug regimen. In this process there is a reduction in viral replication leading to preservation of the CD4+ T cell population by reducing direct loss of infected CD4+ T cells via viral cytopathic effects. There is also reduced killing of infected CD4+ T cells by virus-specific CD8+ cytotoxic T cells.
Soon after initiation of antiretroviral therapy there is also T cell redistribution which occurs due to the reversal of CD4+ lymphocyte homing and sequestration in lymph nodes and redistribution of these cells to the peripheral blood.
2) CD4+ T cell decline and impaired proliferation
Is a decline in CD4 counts, despite viral suppression, and is likely due to drug toxicity. The decrease in immune activation is also taken to be a result of viral replication inhibition leading to a decrease in adhesion molecules at the surface of CD4+ T-cells that are responsible for retaining them with seconfary lymphoid organs. These CD4+ T-cells then end up being released into the periphery.
To understand what has most likely occurred in this patient, it is important to understand that Tenofovir (TDF) impairs T cell proliferation due to inhibition of purine nucleoside phosphorylase (PNP) by TDF mono- and diphosphates and that PNP normally metabolizes cellular inosine monophosphate (IMP) as part of the purine salvage pathway. Inhibition of PNP therefore prevents the breakdown of IMP and promotes the biosynthesis of GTP. Didanosine (ddI) is a substrate competitor for PNP and also results in more cellular IMP. Large amounts of GTP are then synthesised and converted to dGTP by cellular ribo nucleotide reductase (RNR) which is controlled by negative feedback mechanisms. Since RNR produces dNTP’s required for DNA synthesis, cell division is impaired and cells are unable to proliferate.
The combination of ddI and TDF is thus associated with higher intracellular levels of ddI because of the inhibition of PNP, preventing the degradation of ddI and promotes the formation of chain-terminating triphosphates. These cause mitochondrial toxicity by inhibition of mitochondrial DNA polymerase gamma. More chain-terminators and fewer dNTP’s in the mitochondrion severely impairs DNA synthesis and promotes apoptosis.
Mitochondrial toxicity
NRTI’s such as ddI are imported into the mitochondrion and metabolized by mitochondrial enzymes. The chain-terminating triphosphates produced also act as substrates for mitochondrial DNA polymerase gamma and inhibit mitochondrial DNA synthesis. Higher levels of cellular ddI due to inhibition of purine nucleoside phosphorylase (PNP) by tenofovir (TDF) result in higher levels of chain-terminators in the mitochondrion. In addition, inhibition of PNP by TDF also promotes the biosynthesis of cellular GTP which is imported into the mitochondrion and converted to dGTP by mitochondrial ribo nucleotide reductase (RNR). By negative feedback mechanisms, RNR is inhibited by dGTP and prevents dNTP production required for DNA synthesis. More chain-terminators and fewer dNTP’s in the mitochondrion severely impairs DNA synthesis and promotes apoptosis.
Didanosine and Tenofovir metabolism
Cellular enzymes metabolize tenofovir (TDF) and generate mono- and diphosphates which are both inhibitors of purine nucleoside phosphorylase (PNP). PNP normally metabolises cellular inosine monophosphate (IMP) while ddI is a substrate competitor against IMP because it is also a substrate of PNP. Inhibition of PNP leads to higher intracellular levels of ddI which are converted to dideoxy-ATP (ddATP) by cellular enzymes. HIV replication is inhibited by ddATP and tenofovir diphosphate, however mitochondrial polymerase gamma is also inhibited by ddATP and results in higher levels of mitochondrial toxicity.
dGTP synthesis
It is thought that tenofovir (TDF) and ddI co-administration may reduce T cell proliferation as a result of the inhibition of purine nucleoside phosphorylase (PNP) by TDF mono- and diphosphates as well as PNP substrate competition with ddI. PNP deficiency is known to cause an absence of T cells seen in certain forms of severe combined immunodeficiency (SCID) with genetic disorders in PNP while impairment of cell division by PNP inhibition is due to excessive GTP biosynthesis because of a lack of degradation of inosine monophosphate (IMP) by PNP. Large amounts of GTP are converted to dGTP by ribonucleotide reductase (RNR) and dGTP inhibits RNR preventing the synthesis of dNTP’s needed for DNA replication. GTP is also imported into the mitochondrion where mitochondrial RNR converts it to dGTP causing a lack of synthesis of dNTP’s and inhibition of mitochondrial DNA synthesis.
3) CD4+ T cell recovery
Occurs from the change in regimen and hence respite in CD4+ T cell toxicity, resulting in increased CD4 counts and continued immune restoration.
In this case the removal of ddI from the drug regimen reduces competition for PNP substrates such as inosine monophosphate and reduces excessive mitochondrial toxicity due to ddI derived chain-terminating triphosphates.
Download images for this case
Final Outcome
- The patient has gained weight, maintained viral suppression and had no clinical evidence of any opportunistic infections while on the second regimen of TDF, AZT and EFV.
- Her CD4 count has remained stable and above baseline.
- She has continued to take cotrimoxazole and multivitamins.
- No other medicine has been prescribed since starting HAART.
Download images for this case
References
Barreiro P et al. (2006). Suboptimal CD4 gains in HIV-infected patients receiving didanosine plus tenofovir. J Antimicrob Chemother, May;57(5):806-9.
Corbeau, P. and Reynes, J. (2011) “Immune reconstitution under antiretroviral therapy: the new challenge in HIV-1 infection,” Blood, 117(21), pp. 5582–5590.
Download images for this case
Evaluation – Questions & answers
What is the diagnosis?
What are the processes that occur during the immune reconstitution process following initiation of antiretroviral therapy?
- Increase in absolute CD4+ T cell counts
- Reduction in viral replication which protects CD4+ T cells from viral infection and limits virus production by infected cells.
- Reduced killing of infected CD4+ T cells by virus-specific CD8+ cytotoxic T cells
- Redistribution of CD4 cells from lymphoid tissue into the periphery
What is the action of Tenofovir and Didanosine?
Didanosine (ddI) is a substrate competitor for PNP thus also resulting in more cellular IMP. Large amounts of GTP are then synthesised and converted to dGTP by cellular ribonucleotide reductase (RNR) which is controlled by negative feedback mechanisms. Since RNR produces dNTP’s required for DNA synthesis, cell division is impaired and cells are unable to proliferate.
How do Tenofivir and Didanosine contribute to CD4+ T Lymphocyte decline
How do Didanosine and Tenofovir cause mitochondrial toxicity
Download images for this case



