





- Patient presentation
- History
- Differential Diagnosis
- Examination
- Investigations
- Discussion
- Treatment
- Final Outcome
- References
- Evaluation - Questions & answers
- MCQ
Patient presentation
A 24 year old female, presented with a 6 day history of progressive weakness of her lower limbs and since this morning has been unable to walk due to complete paralysis.
Acknowledgement
This case study was kindly provided by Dr Monica Mercer, MBChB from Immunopaedia
History
Two weeks ago she had an episode of gastroenteritis with watery profuse diarrhoea lasting 3 days. She had been treated by her local GP who had sent a stool sample for microscopy, culture and sensitivity, which came back as Campylobacter jejuni.
Past medical history
- Previously well, no chronic disease, no previous admissions.
- HIV negative
Past surgical history
- Tonsillectomy age 7
Family history
- Mother has hypertension on treatment and reports good control.
- No positive family history for any other chronic diseases.
Social history
- Lives with her family (mother, father, 3 sisters)
- Full time student
- Non- smoker
- No alcohol
Differential Diagnosis
- Hyperkalaemia
- Hypokalaemia
- Hypophosphataemia
- Vitamin B12 deficiency
- Multiple sclerosis
- Guillain-Barre syndrome
- Spinal cord compression
Examination
On admission
Anxious but otherwise healthy looking, well nourished young woman, awake and alert and able to communicate giving a thorough history.
Vitals
- Blood pressure 139/80, higher then usual
- Heart rate 84
- Respiratory rate 18
- Temperature 36C
- Oxygen saturation 97% on room air
General
- No signs of dehydration
- No jaundice
- No pallor
- No oedema
- No stigmata of HIV
Respiratory
- No signs of respiratory distress
- Chest clear, good air entry bilaterally
Cardiovascular
- All pulses present, normal rate, rhythm and character
- BP higher then normal for this patient
- No raised JVP
- Apex beat normally placed
- No palpable P2, no parasternal heave, no thrills
- S1, S2 present, no murmurs
Abdomen
- Soft and non tender
- Bowel sounds present
Neurological
- Higher function intact, orientated to person, place and time
- No signs of neck stiffness or meningism
Gait
- Could not be assessed as patient unable to walk due to paralysis of lower limbs
Cranial nerves
- No cranial nerve involvement
Power
- Lower limbs- left and right, 0/5
- Upper limbs- left and right, 5/5
Tone
- Lower limbs- left and right, hypotonia
- Upper limbs-left and right, normal tone
Reflexes
- Lower limbs 0/4, areflexic
- Upper limbs 2/4
- Down going plantars on left and right
- No primitive reflexes elicited
Sensation
- Intact globally
Vibration sense and proprioception
- Intact globally
Day 1 post admission
The patient was admitted to the neurological ward and her symptoms continued to worsen. She started to complain of dysesthesias of her fingers and her upper limbs were now showing weakness, both left and right sides were assessed with power 3/5
She also complained that her face felt weak and she was slurring her words, unable to articulate properly. On examination she appeared to have a Bell’s palsy. She also developed dysphagia and was also unable to swallow without choking and coughing while being fed her breakfast. ICU consultants were called to assess her and it was decided that she should be immediately intubated because of the likelihood of impending respiratory compromise.
Investigations
| Examination | Value | Normal Limits |
|---|---|---|
| WBC | 4.0999999999999996 | (4-12 x109/L) |
| HB | 12.8 | (12.1-15.2 g/L) |
| Platelets | 291 | (140-450 x109/L) |
| CRP | 66 | (0-8mg/l) |
| NA | 133 | (135-147 mmol/L) |
| K | 4.5999999999999996 | (3.3-5.0 mmol/L) |
| CL | 100 | (99-103 µmol/L) |
| C02 | 26 | (18-29 mmol/L) |
| Urea | 7 | (2.5-6.4 mmol/L) |
| Creatinine | 120 | (62-115 mmol/L) |
| Total protein | 111 | (60-80g/L) |
| Albumin | 36 | (35-50g/L) |
| Corrected Calcium | 2.4700000000000002 | (2.1-2.6mmol/l) |
| Phosphate | 1.45 | (1.0-1.5 mmol/l) |
| Magnesium | 0.89 | (0.8-1.3) |
| HIV Elisa | non reactive | |
| CSF | ||
| Glucose | 3.2 | (3-5) |
| Protein | 1.33 | ( |
| lymphocytes | 6 | 0 |
| Neutrophils | 0 | 0 |
| Erythrocytes | 1 | 0 |
| Cryptococcus latex and Indian Ink | negative | |
| Thyroid stimulating hormone | 10.3 | (9-30) |
| Free T4 | 3.13 | (0.5-12.5) |
- Nerve conduction studies showed absent conduction in the lower limbs.
- MRI was suggestive of nerve root enhancement.
Introduction
This case study explores Guillain-Barre syndrome (GBS), which is classically thought of as a rapidly progressive acute polyneuropathy. Several pathologic and aetiologic subtypes exist. These include the most commonly identified subtype – acute inflammatory demyelinating polyneuropathy (AIDP) which presents with loss of motor function, acute motor axonal neuropathy (AMAN) which affects motor function in children and acute motor-sensory axonal neuropathy (AMSAN) which affects both motor and sensory function. Most importantly, the syndrome is generally preceded by a respiratory or gastrointestinal infection, the most common associated organism being the diarrhoea-causing bacteria Campylobacter jejuni. However, Haemophilus influenzae, Mycoplasma pneumoniae, CMV, HIV and EBV have also been associated with GBS. The risk of developing GBS may also occur following vaccination.
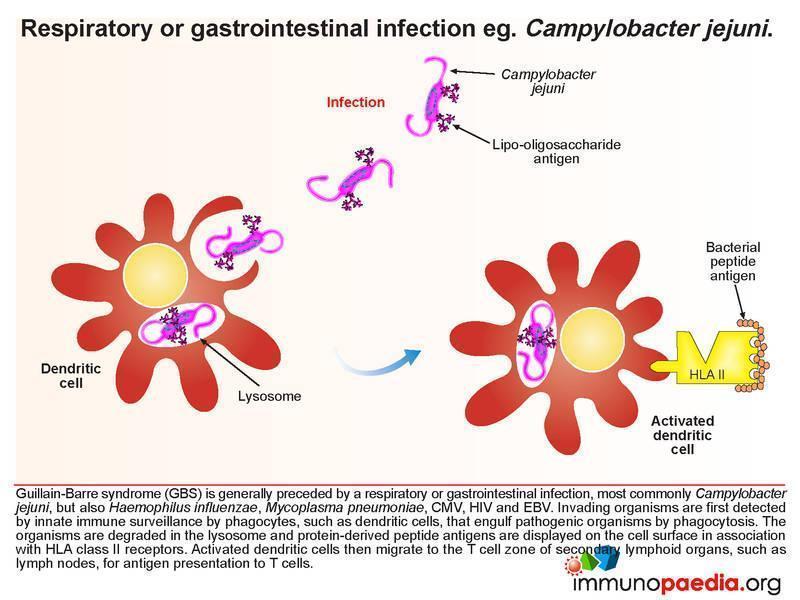
Immunopathology of GBS
The important immunological concept being explored in this case study is molecular mimicry, where it is thought that cross-reactivity of antibodies directed towards the original infecting organism with epitopes on gangliosides found on neurons and myelin occurs. These antibodies cross the blood-nerve barrier and mediate immune attack on peripheral nerves. Inflammatory responses produce cytokines that upregulate vascular integrins to facilitate immune cell infiltration as well as increasing the permeability of capillary endothelial cell linings, which leads to increased diffusion of proteins, particularly immunoglobulins, across the blood-nerve barrier. In GBS, cerebrospinal fluid samples often contain elevated levels of protein but low numbers of cells, as presented in this case.
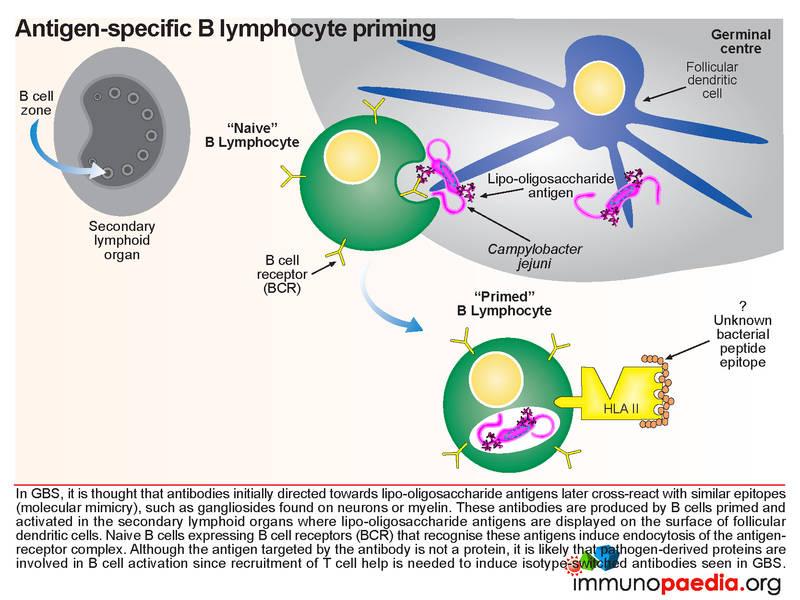
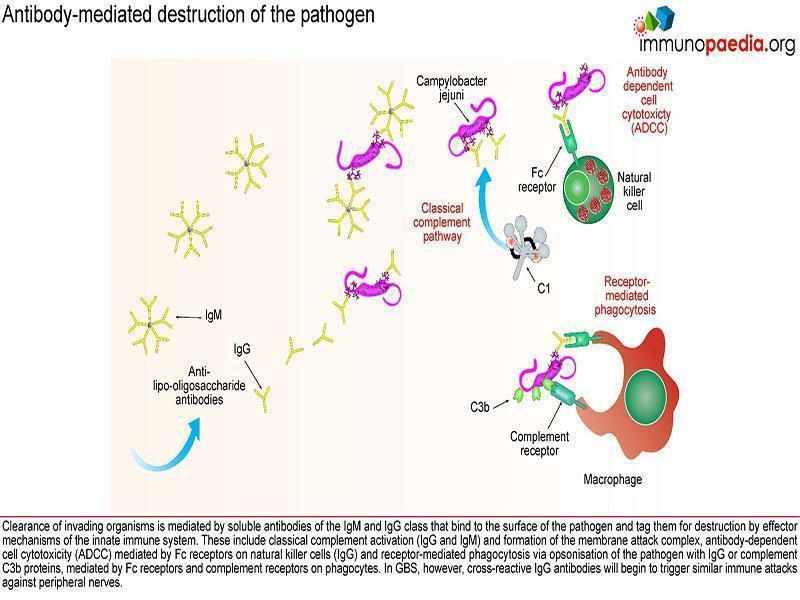
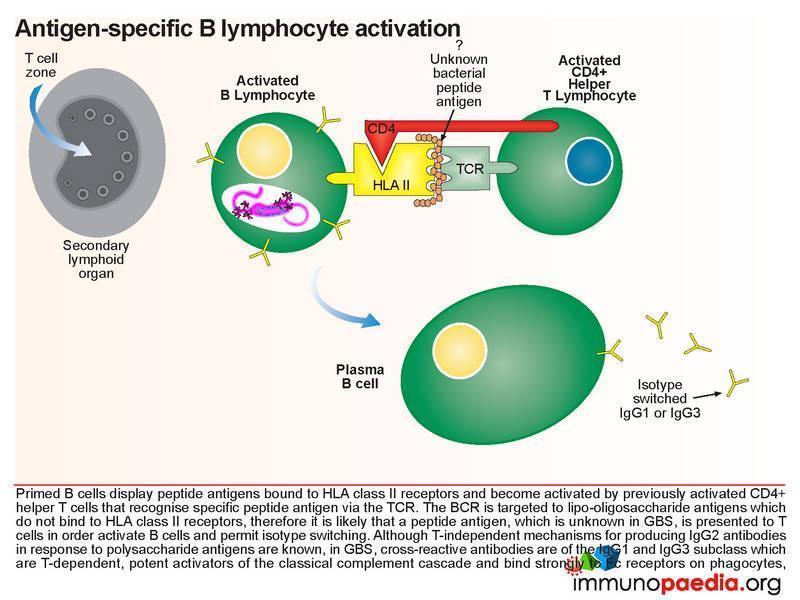
Initially, antibodies are directed towards lipo-oligosaccharides which are non-protein containing antigens. Antibodies are produced by B cells that are primed and activated in the secondary lymphoid organs where the lipo-oligosaccharide antigens are displayed on the surface of follicular dendritic cells. Naive B cells expressing B cell receptors recognise these antigens and induce endocytosis of the antigen-receptor complex. Although the target antigen is not a protein, it is likely that pathogen-derived protein components are involved in B cell activation since cross-reactive antibodies in GBS have been identified as belonging to the IgG1 and IgG3 class. For polysaccharide antigens, it is known that T-independent isotype switching occurs from IgM to IgG2, whilst T-dependent antibody production is known to occur in the presence of peptide antigens displayed by HLA class II receptors. The CD4+ helper T cells then activate B cells, which produce IgM and then later isotype switch to IgG1 and IgG3. These isotypes of antibodies bind well to Fc receptors found on macrophages, for example, and are potent activators of the classical complement cascade, which assists with the clearance of infecting organisms. Such clearance is achieved via three main mechanisms: (a) IgG antibody-dependent cell cytotoxicity (ADCC) mediated by natural killer cells; (b) receptor-mediated phagocytosis via opsonisation of the pathogen with IgG or complement C3b proteins and (c) activation of the classical complement cascade by IgM and IgG and formation of the membrane attack complex. In GBS, however, anti-ganglioside IgG antibodies trigger similar immune attacks involving complement activation and phagocytic activity against peripheral nerves resulting in demyelination of the nerve sheaths or damage to nerve axon or motor end-plates.
 Demyelination of nerve cellsLooking at this in more detail the most common subtype of GBS: acute inflammatory demyelinating polyneuropathy (AIDP) – the demyelination process is associated with infiltration of macrophages and CD4+ helper T cells into peripheral nerve tissue (usually motor neurons). It is thought that anti-myelin IgG antibodies, possibly targeting a ganglioside antigen, mediate destruction of myelin via the classical complement cascade and formation of the membrane attack complexes, as described above. This also activates macrophages expressing Fc receptors for antigen-bound IgG and complement receptors for surface bound C3b that facilitate stripping of myelin. Further enhancement of macrophage activity is also thought to be due to secretion of pro–inflammatory cytokines from CD4+ helper T cells. The resulting destruction and infiltration of myelin by immune cells and membrane attack complexes gives rise to the clinical manifestation of classical rapidly progressive acute polyneuropathy. The patient exhibits all the symptoms described in our case study; progressive symmetric ascending muscle weakness, paralysis and hyporeflexia with or without sensory or autonomic symptoms. Although our patient was intubated and ventilated in time by the clinical team, her muscle weakness was causing respiratory failure, which would have been fatal. The axon demyelination was most likely occurring primarily in peripheral nerves and spinal roots but may also involved her cranial nerves.
Demyelination of nerve cellsLooking at this in more detail the most common subtype of GBS: acute inflammatory demyelinating polyneuropathy (AIDP) – the demyelination process is associated with infiltration of macrophages and CD4+ helper T cells into peripheral nerve tissue (usually motor neurons). It is thought that anti-myelin IgG antibodies, possibly targeting a ganglioside antigen, mediate destruction of myelin via the classical complement cascade and formation of the membrane attack complexes, as described above. This also activates macrophages expressing Fc receptors for antigen-bound IgG and complement receptors for surface bound C3b that facilitate stripping of myelin. Further enhancement of macrophage activity is also thought to be due to secretion of pro–inflammatory cytokines from CD4+ helper T cells. The resulting destruction and infiltration of myelin by immune cells and membrane attack complexes gives rise to the clinical manifestation of classical rapidly progressive acute polyneuropathy. The patient exhibits all the symptoms described in our case study; progressive symmetric ascending muscle weakness, paralysis and hyporeflexia with or without sensory or autonomic symptoms. Although our patient was intubated and ventilated in time by the clinical team, her muscle weakness was causing respiratory failure, which would have been fatal. The axon demyelination was most likely occurring primarily in peripheral nerves and spinal roots but may also involved her cranial nerves.
 Axonal or end-plate terminal damage
Axonal or end-plate terminal damage
Probably unlikely in our patient, but is worth noting, is a less common variant of GBS involving immunological damage to the axon or motor-nerve-end-plate terminals, mediated by anti-ganglioside IgG antibodies. The symptoms and severity depend on the type of ganglioside targeted since the membrane distribution of these molecules on nerve cells varies. Similar to AIDP GBS, immunological damage to nerve cells is caused by activation of the classical complement cascade and formation of the membrane attack complex and by infiltration of macrophages expressing Fc receptors for IgG and complement receptors for C3b that initiate phagocytic processes.
Download images for this case
Treatment
The patient in our case study was treated with intravenous immunoglobulin (IVIG) 0.4g/kg/24hr IVI for 5 days. IVIG is derived from fractionated, purified human plasma collected from a large pool of multiple donors. The exact mechanism of action is not known but several mechanisms are thought to be involved which include the blockage of macrophage receptors, the inhibition of antibody production, the inhibition of complement binding and the neutralization of pathologic antibodies. IVIG resulted in successful treatment and the patient showed signs of recovery.
An alternative therapy is plasmapharesis. This mechanism is used to remove IgG from the serum. During plasmapheresis, blood is initially taken out of the body through a needle or previously implanted catheter. Plasma is then removed from the blood by a cell separator, centrifuged or filtered to separate the plasma from blood cells. The blood cells are then returned to the patient with additional fresh frozen donor plasma, albumin or saline with added proteins. Exchanges are performed according to clinical presentation, usually patients receive 3-5 exchanges over a 2 week period.
Studies have shown that both treatments are equally effective, while combined treatment has no added benefit. Treatment is based on disease severity and rate of progression, as well as on the length of time between the condition’s first symptom and current presentation. Risk of a thrombotic event associated with IVIGs should be carefully monitored.
Download images for this case
Final Outcome
Our patient was extubated following 14 days of ventilation and there was a gradual clinical improvement over 6 weeks. The patient received intense physiotherapy during this time and her symptoms continued to resolve and was able to walk again, although with some residual weakness remaining in her lower limbs. She has a good prognosis and is expected to make a full recovery within 18 months.
Download images for this case
References
Gupta A et al. (2010). “Guillain-Barre Syndrome rehabilitation outcome, residual deficits and requirement of lower limb orthosis for locomotion at 1 year follow-up”. Disabil Rehabil. 24 March
Jacobs BC et al. (2008). “Subclass IgG to motor gangliosides related to infection and clinical course in Guillain-Barré syndrome”. J Neuroimmunol. 4 January
Jacobs BC et al. (1996). “Campylobacter jejuni infections and anti-GM1 antibodies in Guillain-Barré syndrome”. Ann Neurol. August. 40(2): 181-7.
Kenichi Kaida et al. (2009). “Antiganglioside antibodies and their pathophysiological effects on Guillain–Barr´e syndrome and related disorders—A review” Glycobiology vol. 19 no. 7 pp. 676–692
Pieter A van Doorn, Liselotte Ruts, Bart C Jacobs. (2008). “Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome”. Lancet Neurol. 7: 939–50
Download images for this case
Evaluation – Questions & answers
What is the diagnosis?
What is thought to be the cause of this syndrome?
Can B cells differentiate into plasma cells and isotype switch without T cell help?
Which organisms are most commonly associated with this syndrome?
What are the immunopathological events leading to GBS?
- Antibodies produced by B cells that are primed and activated in the secondary lymphoid organs by the infecting organism.
- Activation of the classical complement cascade occurs to clear the pathogenic organism.
- Due to molecular mimicry, where IgG targeting lipo-oligosaccharide epitopes on the infecting organism cross-react with similar ganglioside-looking epitopes on nerves and myelin, the antibodies trigger complement-mediated cytotoxicity and phagocytic attack against the myelin sheath surrounding peripheral nerves or damage to the nerve axon or end-plates.
- There is demyelination of the nerve sheaths and a rapidly progressive acute polyneuropathy or loss of nerve function due to damage to the axon or end-plate.
Download images for this case
Multiple Choice Questions
Earn 1 HPCSA or 0.25 SACNASP CPD Points – Online Quiz
Download images for this case



