





- Patient presentation
- History
- Examination
- Diagnostic Hypotheses
- Investigations
- Discussion
- Differential Diagnosis
- Treatment
- Final Outcome
- Conclusion
- References
- Evaluation - Questions & answers
- MCQs
Patient Presentation
Emilio, a 60 years-old, bank accountant, presented with a complaint of erythematous patches covered with fragile blisters that were evolving into erosions.
Acknowledgement
This case was provided by Dr. Marion Carrette (PharmD, Department of Immunology and Biotherapy, Rouen University Hospital, France) and Prof. Olivier Boyer (MD, PhD, Head of the Department of Immunology and Biotherapy, Rouen University Hospital, France). The authors would like to thank Prof. Vivien Hébert (MD, PhD, Department of Dermatology, Rouen University Hospital, France), Dr. Fabienne Jouen (MD, Department of Immunology and Biotherapy, Rouen University Hospital, France), and Prof. Victoria Werth (MD, Department of Dermatology, University of Pennsylvania, Philadelphia, USA) for their critical reading of this case study.
History
Two months ago, Emilio started to develop painful oral lesions which affecetd food intake. Small erythematous plaques concomitantly appeared on Emilio’s scalp. Flaccid blisters were visible on the plaques, leading to skin detachment and erosions. Soon after, the plaques on the scalp began to expand and new plaques appeared on his neck, chest and back.
Past medical history
- Hypercholesterolemia
- No recent illness
Past surgical history
- Hallux valgus surgery for the left foot at 45
Family history
- Father died of lung cancer at 62 years-old
- Mother has Grave’s disease (autoimmune thyroiditis)
Travel history
- No recent travel
Social history
- Married
- 2 children (29 and 26 years old)
- Bank accountant
Medication
- Atorvastatin (lowers blood levels of cholesterol)
- No recent change in medication
Examination
Vitals
- Oxygen saturation: 96%,
- Blood pressure: 130/80 mmHg,
- Temperature: 37.1°C
- Heart rate: 85/min
General
- Height: 1m78
- Weight: 85 kg with a loss of 3 kg in the last 2 months
- Visual analog scale for pain assessment: 5 (moderate pain) to 7 (severe pain) when eating and swallowing food
- No sign of lymphadenopathy
Cardiovascular
- Normal heart sounds
- All pulses present
Respiratory
- No dyspnea (labored breathing)
- Normal auscultation
- Respiratory rate: 18/min
Abdomen
- Normal
Cutaneous
- Multiple fragile blisters were noted on the scalp, neck, chest and top of the back associated with erosions on more than 5% of the total body surface
- Positive Nikolsky’s sign
- Mucous membrane erosions were present on the oral mucosa, the tongue, the floor of the mouth and the posterior pharynx
- PDAI score (see definition below): 46
Diagnostic Hypotheses
- Mucous Membrane Pemphigoid
- Pemphigus Vulgaris
- Bullous Pemphigoid
- Linear IgA Bullous Dermatosis
- Epidermolysis Bullosa Acquisita
- Erythema Multiforme
- Drug-induced Bullous Disorders
Investigations
| Examination | Value | Normal limits |
|---|---|---|
| Leucocytes (white blood cells) | 8.2 | (4-12 x109 /L) |
| Hemoglobin | 14.1 | (12.1-15.2 g/L) |
| Platelets | 320 | (140-450 x109 /L) |
| CRP | 5 | (0-8 mg/l) |
| Sodium | 136 | (135-147 mmol/L) |
| Potassium | 4.5 | (3.3-5.0 mmol/L) |
| Chloride | 100 | (99-103 μmol/L) |
| Urea | 6.2 | (2.5-6.4 mmol/L) |
| Creatinine | 75 | (62-115 mmol/L) |
| Total protein | 65 | (60-80 g/L) |
| Albumin | 40 | (35-50 g/L) |
| Antinuclear autoantibodies | Negative | |
| Indirect immunofluorescence (IIF) on human split skin | Negative | |
| IIF on monkey esophagus | Basal membrane zone: negative
Intercellular spaces: positive, titer 1/320 |
|
| Anti-Dsg3 antibodies | 180 | (<20 UA/ml) |
| Anti-Dsg1 antibodies | 160 | (<20 UA/ml) |
| Anti-BP180 antibodies | 2 | (<20 UA/ml) |
| Anti-BP230 antibodies | 6 | (<20 UA/ml) |
| Anti-collagen VII antibodies | 1 | (<20 UA/ml) |
Skin biopsies:
- Histology: suprabasal acantholysis was observed
- Direct Immunofluorescence (DIF): interkeratinocytes deposition of IgG and C3 in the lower epidermis
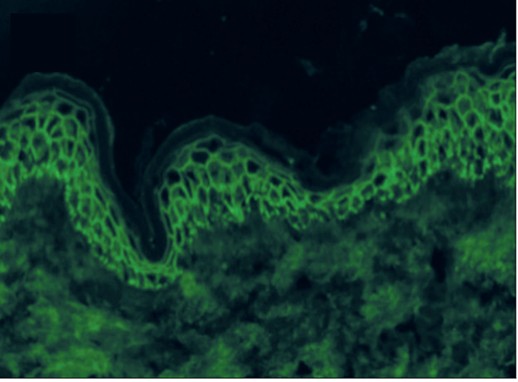
Figure 1: Fishnet pattern deposition of IgG in Emilio’s epidermis by direct immunofluorescence (DIF)
Discussion
The clinical presentation and biological findings are consistent with the diagnosis of pemphigus vulgaris (PV), a rare autoimmune bullous dermatosis characterized by mucosal involvement in 90% of patients. The disease usually occurs between the ages of 45 and 65 years and a slight female predominance has been reported, with M:F ratios between 1:1.1 – 1:1.7. Its incidence differs considerably between populations, ranging from 0.6/million/year in Switzerland to 32/million/year in Israel, with strong HLA association with the DRB1*04:02 and DQB1*05:03 alleles1.
Physiopathology
Pemphigus vulgaris (PV) is an autoimmune blistering disorder characterized by the production of pathogenic autoantibodies targeting desmosomal adhesion molecules, primarily desmoglein 3 (Dsg3) and desmoglein 1 (Dsg1) (Figure 2). The pathophysiology of PV involves a cascade of events that disrupt the normal function of desmosomes, which are crucial for maintaining the structural integrity of the epidermis and mucous membranes. Autoantibodies against Dsg3 and Dsg1, primarily of IgG1 and IgG4 classes, bind to the extracellular domains of these desmosomal proteins, triggering intraepidermal acantholysis, a hallmark feature of PV2. This binding leads to the formation of pathogenic immune complexes and activation of downstream signaling pathways, which ultimately results in the disruption of desmosome-mediated cell adhesion. Consequently, keratinocytes lose their cohesive interactions, leading to the formation of suprabasal blisters and erosions within the epidermis and mucous membranes. The disruption of epithelial integrity not only results in the characteristic clinical manifestations of PV but also predisposes patients to secondary infections and systemic complications.
The pathogenic effects of anti-desmoglein antibodies have been demonstrated through experimental models using patient’s purified anti-Dsg3 IgG and the monoclonal anti-Dsg3 antibody AK23, showing reduction of Dsg3 membranous expression and loss of cell adhesion on HaCaT cells (immortalized human keratinocytes) in vitro, in human skin ex vivo and in neonatal mice in vivo3. Passive transfer of splenic B and T cells from Dsg3-/- mice immunized with a recombinant Dsg3 protein (recognized here as a non-self protein, favoring the development of anti-Dsg3 antibodies) into RAG-/- mice (animals without an adaptive immune system but with normal Dsg3 expression) induces a pemphigus phenotype4. In patients, decreasing autoantibody levels correlate with a decrease in clinical activity. Moreover, placental transfer of anti-desmoglein IgG from the mother can be associated with neonatal PV in the fetus.
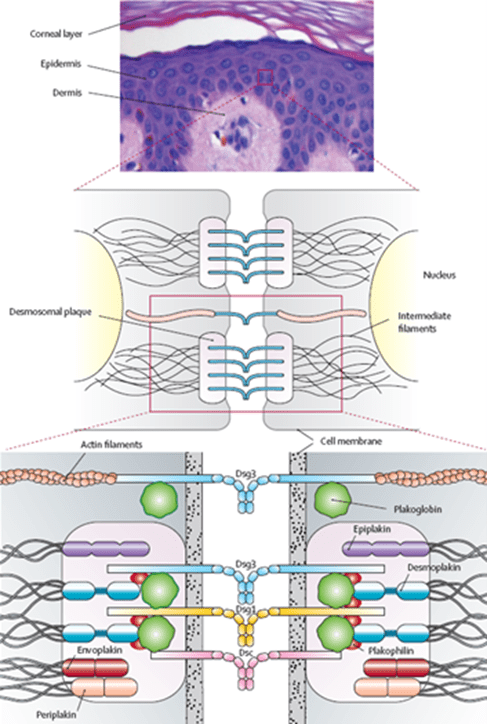
Figure 2: Schematic representation of the epidermal desmosome1. Dsg3: desmoglein 3; Dsg1: desmoglein 1; Dsc: desmocollin.
In PV, pathogenic antibodies are mainly produced by short-lived plasmablasts (SLPB) produced through an extrafollicular response, in contrast to long-lived plasma cells (LLPC) that originate from activated B cells in germinal centers of secondary lymphoid organs (Figure 3)5. Anti-Dsg3 and anti-Dsg1 autoantibody levels are strongly associated with disease activity and their monitoring is useful for the disease follow-up1.
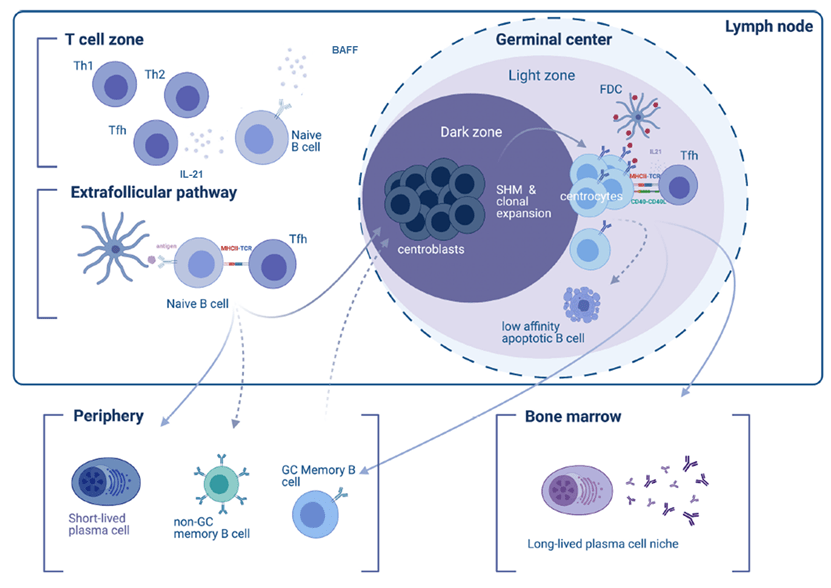
Figure 3: The cellular stages of antibody-secreting cells (ASC)6. Th, helper T cells; Tfh, follicular helper T cells; SHM, somatic hypermutations; FDC, follicular dendritic cells; GC, germinal centers.
Clinical presentation and classification
Two clinical variants of PV exist: mucosa-dominant PV and mucocutaneous PV. In most patients, the first lesions generally appear in the oral cavity and nearly all patients will develop oral lesions at some point in the disease. Erosions can be found on the buccal mucosa, palate, tongue, inner side of the lips and gingiva. These lesions are often associated with pain, which can interfere with eating and sometimes leading to rapid weight loss. Other mucosal surfaces are less frequently involved, such as nasal mucosa, esophagus, urethra, glans penis, vulva and anus. In the mucocutaneous variant, skin lesions can arise along with mucosal lesions or later1.
For the management of PV, disease severity and treatment response are assessed using the Pemphigus Disease Area Index (PDAI) clinical scoring systems 7,8. PDAI is a comprehensive scoring system used to evaluate the extent and severity of pemphigus vulgaris lesions. It assesses the involvement of various body areas (skin, mucous membranes), the type and extent of lesions, and the presence of subjective symptoms such as pain and pruritus. PDAI scores range from 0 to 250, with higher scores indicating more severe disease activity. In Emilio’s case, is PDAI was 46 (Figure 4, Table 1).
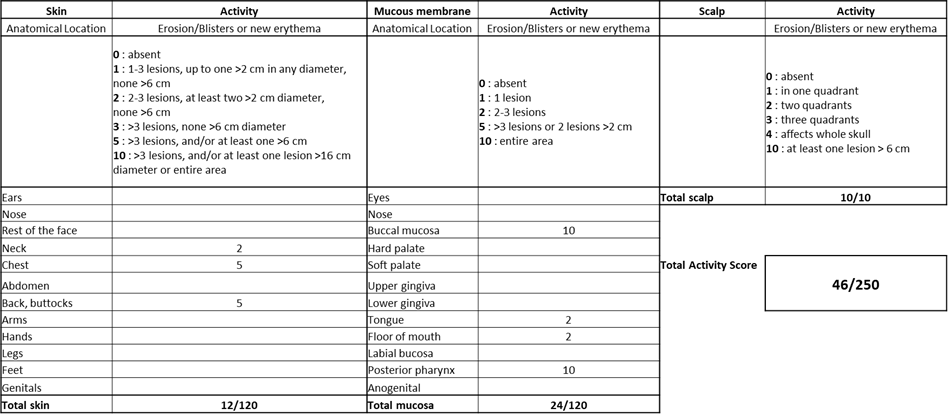
Figure 4: Emilio’s PDAI score detail.
| Disease activity | PDAI |
| Mild | <15 |
| Moderate | 15-45 |
| Severe | >45 |
Table 1: PDAI scores and disease activity8.
Thus, Emilio disease activity is classified as severe PV with a PDAI of 46.
Differential Diagnosis
- Mucous Membrane Pemphigoid (MMP): MMP is another autoimmune blistering disease primarily affecting mucous membranes, including the oral cavity, eyes, and genitalia. It is characterized by subepithelial blistering and linear deposition of IgG and/or IgA antibodies along the basement membrane zone. MMP can present with erosions and scarring involving the oral mucosa, resembling PV.
- Bullous Pemphigoid (BP): BP is an autoimmune blistering disease characterized by tense subepidermal blisters that predominantly affect elderly individuals. While oral involvement is less common in BP compared to PV and MMP, it can present with erosions and blisters in the oral mucosa. Diagnosis is based on histopathology demonstrating subepidermal blistering and direct immunofluorescence shows linear deposition of IgG and/or C3 along the basement membrane zone. Salt split testing skin shows antibodies on the roof of the blister cavity, allowing distinction of BP from Epidermolysis Bullous Acquista (EBA). ELISA testing for anti-Bullous Pemphigoid (BP) 180 and anti-BP 230 autoantibodies is normally also performed for diagnosis.
- Linear IgA Bullous Dermatosis (LABD): LABD is an autoimmune blistering disease characterized by linear deposition of IgA along the basement membrane zone. It can present with tense bullae and erosions involving the skin and mucous membranes, including the oral cavity. Diagnosis is confirmed by DIF demonstrating linear IgA deposition.
- Epidermolysis Bullosa Acquisita (EBA): EBA is a rare autoimmune blistering disease characterized by autoantibodies targeting type VII collagen, resulting in subepidermal blistering. While oral involvement is less common in EBA, it can present with erosions and blisters affecting the oral mucosa, similar to other autoimmune blistering diseases. Diagnosis is confirmed by histopathology and immunofluorescence studies.
- Erythema Multiforme (EM): An acute, self-limited hypersensitivity reaction characterized by targetoid lesions and mucosal involvement. EM may present with bullae and erosions resembling PV, particularly in severe cases, but typically lacks the chronicity and autoantibody involvement seen in PV. The morphology of skin lesions is typically different from PV.
- Drug-induced Bullous Disorders: Certain medications, such as antibiotics (e.g., sulfonamides), anticonvulsants (e.g., phenytoin), and diuretics (e.g., furosemide), can induce blistering reactions resembling autoimmune blistering diseases. Differentiation may require careful medication history and cessation of the offending agent.
Treatment
As an initial presentation of severe PV, Emilio has been treated by oral prednisolone at 1mg/kg daily plus the anti-CD20 antibody rituximab (2x 1g intravenously at 15 days of interval)9. Symptomatic treatments for pain management and wound care, including topical emollients and oral analgesics, were also provided10. Before treatment initiation, Emilio was vaccinated against influenza and pneumococcus and had screening labs before treatment with rituximab, as summarized below (Figure 5).
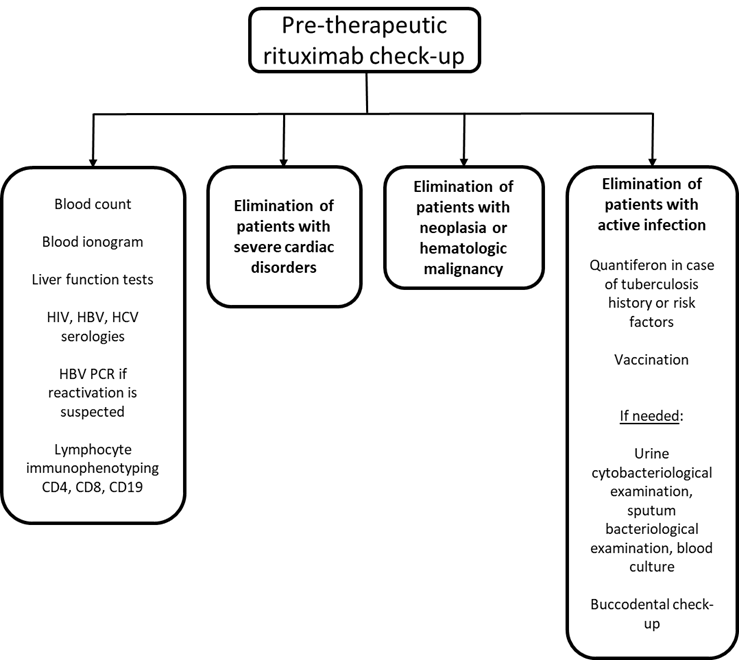
Figure 5: Pre-therapeutic check-up for rituximab.
After one month, Emilio’s clinical status has largely improved with no new blisters and a regression of the erosive post-bullous lesions. His blood test showed a significant decrease in anti-Dsg3 and anti-Dsg1 autoantibody levels, B cell count of 0, but normal levels of total IgG, IgA and IgM associated with protecting levels of IgG in vaccine serology against influenza and pneumococcus (Figure 6). Progressive prednisone dose tapering was initiated with the objective to stop corticosteroids after 4 months of treatment.
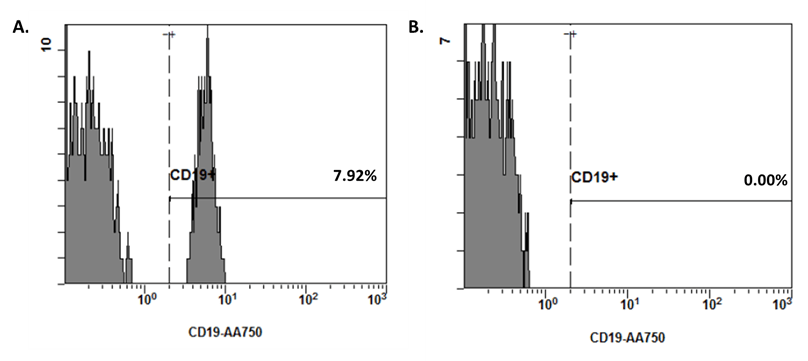
Figure 6: Frequencies of CD19+ B cells among Emilio’s blood lymphocytes before (panel A) and after (panel B) rituximab introduction.
Rituximab for the treatment of autoimmune diseases
Rituximab is a monoclonal chimeric anti-CD20 antibody. CD20 is a pan B-cell marker, expressed from the pre-B cell stage to the mature B cell stage (Figure 7).
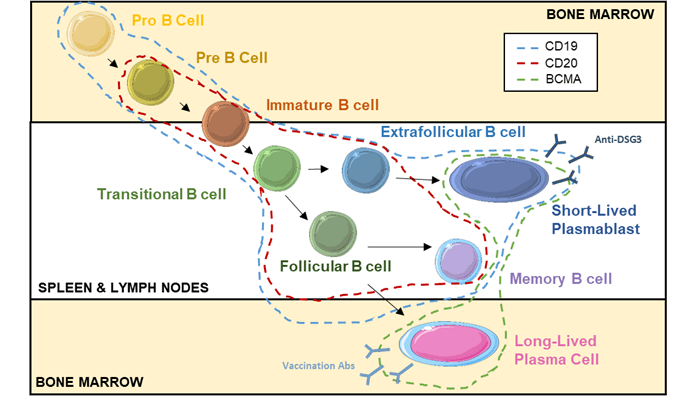
Figure 7: Expression of CD19, CD20 and BCMA along B cell lymphopoiesis and maturation.
Rituximab was first used as an antitumor treatment for B cell malignancies and has revolutionized the management of B cell lymphoma. Given its efficacy in depleting B cells, it has been subsequently evaluated in different autoimmune diseases with varying results. In the management of PV, it has showed impressive results to the point that international guidelines have now recommended it as first–line treatment for moderate and severe PV11. However, it has shown more limited therapeutic efficacy in systemic lupus erythematosus, an autoimmune disease associated with the presence of anti-dsDNA autoantibodies.
Notably, rituximab does not deplete antibody-secreting cells, i.e. plasma cells as they do not express CD20. However, it is particularly effective in diseases where pathogenic antibodies are produced by B cells or short-lived plasmablasts (SLBP) because, due to their short life, SLBP require continuous replenishment from the autoreactive mature B cell pool. This explains why pathogenic autoantibody levels are decreased12. In contrast, as observed in Emilio’s case, treatment with rituximab did not affect the production of vaccine antibodies that originate from long-lived plasma cells (LLPC) that do not express CD20 and can persist for several years. Other autoantibodies such as anti-SSA IgG in systemic lupus erythematosus are also resistant to rituximab therapy (Figure 8)13.
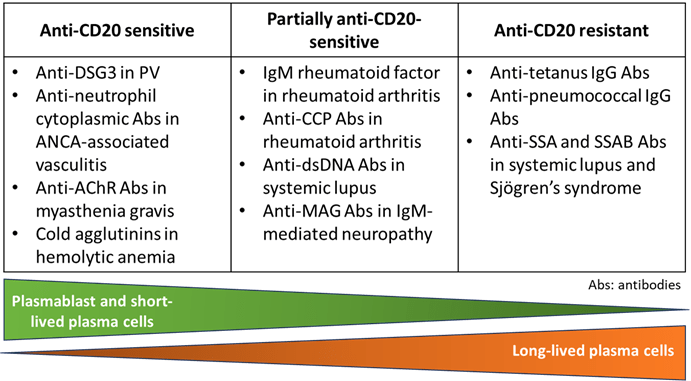
Figure 8: Sensitivity to anti-CD20 treatment in function of the type of antibody-secreting cells (ASC) involved in antibody production. ANCA: antineutrophil cytoplasmic antibodies; AChR: acetylcholine receptor; CCP: cyclic citrullinated peptide; MAG: myelin associated glycoprotein; SSA and SSB: Sjögren’s syndrome-A and B.
The main adverse effects of rituximab are the risk of long-term hypogammaglobulinemia with repeated treatments, late-onset neutropenia and infections14.
Other B-cell depletion strategies
- Belimumab (anti-BAFF): belimumab is a monoclonal antibody used primarily in the treatment of systemic lupus. It works by inhibiting the activity of B-cell activating factor (BAFF), a protein that promotes the survival and activation of B cells, which are responsible for producing autoantibodies. By reducing the activity of BAFF, belimumab helps to decrease the number of abnormal B cells, thereby reducing disease activity and flares in lupus patients. It is typically used in patients who have not responded well to standard therapies. Belimumab is approved for lupus nephritis and is being studied in Sjögren’s syndrome15.
- Daratumumab (anti-CD38): daratumumab is a monoclonal antibody primarily approved for the treatment of multiple myeloma. However, its use is being explored in the context of autoimmune diseases, due to its ability to target CD38, a protein expressed on the surface of plasma cells. By targeting and depleting these plasma cells, daratumumab has shown potential in treating refractory autoimmune hemolytic anemia (AIHA) and other antibody-mediated autoimmune disorders. Clinical trials and case studies suggest that daratumumab may reduce disease activity in these conditions by decreasing the production of harmful autoantibodies, providing a new therapeutic option for patients who do not respond to conventional treatments16.
- CD19 CAR-T cells: CD19 CAR T-cell therapy, initially developed for treating certain types of cancers like B-cell lymphomas and leukemias, is being explored as a novel treatment for autoimmune diseases. This therapy involves modifying a patient’s T cells to express chimeric antigen receptors (CARs) that specifically target the CD19 protein found on the surface of B cells. By targeting and eliminating these B cells, CD19 CAR T-cell therapy aims to reduce the production of autoantibodies and thereby mitigate disease activity. Early clinical studies and trials have shown promising results, particularly in severe, treatment-resistant cases of autoimmune diseases such as systemic lupus. The potential benefits of CD19 CAR T-cell therapy in autoimmunity include long-lasting remission and the possibility of reducing reliance on chronic immunosuppressive medications17.
Final Outcome
Regular follow-up visits were scheduled to monitor clinical response, disease activity, and adverse effects of treatment. Emilio demonstrated significant improvement in his PV lesions one month following rituximab therapy. The number and severity of blisters and erosions decreased, and mucosal involvement resolved substantially. Prednisone dose was tapered over 6 months successfully without disease flares. Anti-Dsg3 and anti-Dsg1 autoantibodies remained negative 3 months after rituximab infusion. Rituximab was repeated 6 months after the first injection (1g intravenously). Follow-up evaluations revealed sustained remission of his disease with no significant adverse effects and B cell counts returned to normal 1 year after the last infusion of rituximab.
Conclusion
PV is a severe chronic autoimmune disease associated with pathogenic anti-Dsg3 and anti-Dsg1 autoantibodies responsible for the formation of blisters and erosions in the mouth mucosa and epidermis. The anti-CD20 antibody rituximab is an effective treatment option for moderate-severe PV or refractory cases as it removes B cells and impairs the pool formation of short-lived plasmablasts responsible for autoantibody production. Close monitoring of clinical response, disease activity, and adverse effects linked to the B cell depletion induced by rituximab are essential in managing patients with PV.
References
- Amagai, M., Karpati, S., Prussick, R., Klaus-Kovtun, V. & Stanley, J. R. Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J. Clin. Invest. 90, 919–926 (1992).
- Schmitt, T. & Waschke, J. Autoantibody-Specific Signalling in Pemphigus. Front. Med. 8, 701809 (2021).
- Amagai, M. et al. Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. J. Clin. Invest. 105, 625–631 (2000).
- Nutt, S. L., Hodgkin, P. D., Tarlinton, D. M. & Corcoran, L. M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 15, 160–171 (2015).
- Zografou, C., Vakrakou, A. G. & Stathopoulos, P. Short- and Long-Lived Autoantibody-Secreting Cells in Autoimmune Neurological Disorders. Front. Immunol. 12, 686466 (2021).
- Hébert, V. et al. Large International Validation of ABSIS and PDAI Pemphigus Severity Scores. J. Invest. Dermatol. 139, 31–37 (2019).
- Boulard, C. et al. Calculation of cut-off values based on the Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) and Pemphigus Disease Area Index (PDAI) pemphigus scoring systems for defining moderate, significant and extensive types of pemphigus. Br. J. Dermatol. 175, 142–149 (2016).
- Joly, P. et al. Updated S2K guidelines on the management of pemphigus vulgaris and foliaceus initiated by the european academy of dermatology and venereology (EADV). J. Eur. Acad. Dermatol. Venereol. JEADV 34, 1900–1913 (2020).
- Murrell, D. F. et al. Diagnosis and management of pemphigus: Recommendations of an international panel of experts. J. Am. Acad. Dermatol. 82, 575-585.e1 (2020).
- Joly, P. et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet Lond. Engl. 389, 2031–2040 (2017).
- Huang, H., Benoist, C. & Mathis, D. Rituximab specifically depletes short-lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proc. Natl. Acad. Sci. U. S. A. 107, 4658–4663 (2010).
- Mahévas, M., Michel, M., Weill, J.-C. & Reynaud, C.-A. Long-lived plasma cells in autoimmunity: lessons from B-cell depleting therapy. Front. Immunol. 4, 494 (2013).
- Athni, T. S. & Barmettler, S. Hypogammaglobulinemia, late-onset neutropenia, and infections following rituximab. Ann. Allergy Asthma Immunol. Off. Publ. Am. Coll. Allergy Asthma Immunol. 130, 699–712 (2023).
- Levy, R. A. et al. 10 Years of belimumab experience: What have we learnt? Lupus 30, 1705–1721 (2021).
- Holzer, M.-T. et al. Daratumumab for autoimmune diseases: a systematic review. RMD Open 9, e003604 (2023).
- Müller, F. et al. CD19 CAR T-Cell Therapy in Autoimmune Disease – A Case Series with Follow-up. N. Engl. J. Med. 390, 687–700 (2024).
Evaluation – Questions & answers
Are anti-desmoglein autoantibodies pathogenic and why?
Yes, the presence of anti-desmoglein antibodies is a hallmark of PV. The autoantibodies bind to desmosomes and are typically responsible for the acantholysis observed in the disease. Arguments in favor of a pathogenic effect of autoantibodies are:
- Efficacy of rituximab in the disease by B cell and short-lived plasmablast depletion.
- Patient’s purified anti-Dsg3 IgG and the monoclonal anti-Dsg3 antibody AK23 lead to reduction of Dsg3 membrane expression and loss of cell adhesion on different models: HaCaT cells in vitro, human skin ex vivo and neonatal mice in vivo.
- In vivo mouse model: transfer of splenocytes from a Dsg3-immunized Dsg3-/- mouse into a RAG-/- recipient produces a pemphigus phenotype.
- Decreasing autoantibody levels in patients correlate with a decrease in clinical activity of the disease.
- Placental transfer of the anti-desmoglein IgG from the mother may induce PV in the fetus.
Are long-lived plasma cells involved in the pathogenesis of Emilio’s disease?
No, B cells and short-lived plasmablasts (SLBP) are responsible for the production of anti-Dsg1 and anti-Dsg3 in PV. Due to the short half-life of SLBP, production of autoantibodies requires continuous replenishment from autoreactive B cells. The latter are killed by rituximab; this is why although not killing the plasma cells themselves, rituximab is efficient to decrease autoantibody production. In contrast, long-lived plasma cells (LLPC) producing IgG vaccination antibodies are not impaired by the rituximab treatment.



