





- Patient presentation
- History
- Differential diagnosis
- Examination
- Investigations
- Discussion
- Treatment
- Final Outcome
- References
- Evaluation - Questions & answers
- MCQs
Patient Presentation
A five-month-old male infant was brought into the emergency department by parents with hypotonicity (reduced muscular tone) and drowsiness that began the evening before. The infant presented with refractory atopic dermatitis and recent weight loss.
Acknowledgement
This case study was provided by Prof. Olivier Boyer (M.D., Ph.D., Head of the Department of Immunology and Biotherapy, Rouen University Hospital, France) and Dr. Audrey Aussy (M.D., Assistant Professor) of the Faculty of Medicine of Rouen, Normandy University, France). The authors would like to thank Prof. Capucine Picard (M.D., Ph.D., Head of the Study Center for Primary Immunodeficiencies, Necker University Hospital, France), Dr. Serge Jacquot (M.D., Ph.D.) and Dr. Aude Marie-Cardine (M.D.) of Rouen University Hospital’s Reference Center for Hereditary Immunodeficiencies, Dr. Benoit Salomon (D.V.M., Ph.D., Head of the Treg biology and therapy research team, INSERM U1135 CIMI, Pitié-Salpêtrière, Paris) for their critical reading of this case study. We are grateful to Nikki Sabourin-Gibbs, Rouen University Hospital, for her help in editing the manuscript.
Partnership
We have partnered with The International Union of Basic and Clinical Pharmacology (IUPHAR) to bring you in-depth information about drugs and pharmacology with links to the Guide to ImmunoPharmacology.
History
Keran was born at 39 weeks since the LMP (last menstrual period) (weight 3.2 kg). Fifteen days after birth, he developed an eczematous rash on his face which was diagnosed as atopic dermatitis. Local application of hydrocortisone partly improved the cutaneous lesions. His parents report the onset of chronic diarrhoea since the age of two months.
The number of diapers used daily has significantly increased in the last few days due to polyuria (excessive excretion of urine). On the morning of presenting to the clinic Keran was unusually sleepy and unable to breastfeed. He was pale and hypotonic.
Past medical history
- Atopic (allergic) dermatitis
Surgical history
- None
Family history
- Two healthy sisters aged 5 years and 7 years
- A brother born 2 years previously, died at the age of 2 months with severe anaemia, thrombocytopenia (decrease in number of platelets in circulating blood) and diarrhoea
Travel history
- None
Social history
- Father is an airline pilot
- Mother is unemployed
- Childcare: mother
Medication
- Local applications of hydrocortisone
Differential diagnosis
- Meningitis
- Head trauma
- Brain tumour
- Disseminated intra-vascular coagulation
- Ketoacidosis (the accumulation of ketone bodies in the blood)
- Dehydration due to chronic diarrhoea
- Adrenal insufficiency
- Immune dysregulation, polyendocrinopathy, enteropathy X-linked disease (IPEX)
(a rare disease linked to the dysfunction of the transcriptional activator FoxP3) - Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) (A subtype of mucocutaneous candidiasis characterised by increased immunoglobulins and
decreased IgA, with progressive decline in parathyroid and adrenal function) - Child abuse
Examination
Vitals
- Heart rate: 98 beats/min
- Temperature: 37.8°C
- Oxygen saturation: 95%
- Respiratory rate: 28 breaths/min
General
- Pale
- Sleepy and hypotonic
- Weight: 5 kg (< -3 SD for age)
- Cervical adenopathy (enlargement of the lymph nodes)
Cardiovascular
- Normal heart sounds
- All pulses present
Respiratory
- Good bilateral air entry
- No crackling sound on the lungs or wheezing
Abdomen
- Not distended
- Enlarged spleen
- Bowel sounds present
Neurological
- Normal pupillary reflexes
- Opens his eyes when feet are stimulated
- Moderate hyper-reflexia (exaggeration of reflexes) on upper and lower limbs
- No sphincter disorder
Dermatological
- Diffuse lesions of atopic dermatitis
- No sign of local infection
- No purpura (a small hemorrhage in the skin)
- Persistence of skin folds after pinching
Follow-up clinical examinations
At admission, Keran was immediately perfused with crystalloids. Urinalysis revealed high urine glucose and ketone levels. Type I diabetes mellitus was diagnosed. Insulin therapy was started intravenously. After a few days, intravenous insulin therapy was changed to basal-bolus insulin regimen. Physicians noted persistence of watery diarrhoea and absence of weight gain. Parenteral nutrition was required. Eczematous rash also persisted in spite of appropriate local treatments.
Investigations
| Examination | Value | Normal limits at 5 months |
|---|---|---|
| WBC | 8.9 | (6.0-17.5 x10 9 cells/L) |
| Neutrophils | 2.6 | (1.0-8.5 x10 9 cells/L) |
| Lymphocytes | 4.2 | (3.4-9.5 x10 9 cells/L) |
| Monocytes | 1.0 | (0.2-1.2 x10 9 cells/L) |
| Basophils | 0 | (<0.11 x10 9 cells/L) |
| Eosinophils | 1.1 | (<0.8 x10 9 cells/L) |
| Hb | 98 | (114-141 g/L) |
| MCV (Mean corpuscular volume) | 95 | (80-96 f/L) |
| Reticulocyte count | 11.2 | (0.8-4% of RBC) |
| Platelets | 235 | (150-400 x10 9 u/L) |
| LDH (Lactate Dehydrogenase) | 735 | (70-250 u/L) |
| Haptoglobin (free serum) | 2 | (27-139 mg/dl) |
| Bilirubin unconjugated | 2.3 | (<1.0 mg/dL) |
| Alanine aminotransferase | 14 | (6-50 U/L) |
| Aspartate aminotransferase | 23 | (20-60 U/L) |
| Alkaline phosphatase | 214 | (110-320 U/L) |
| γ -Glutamyl transferase (GGT) | 52 | (34-263 U/L) |
| INR (international normalized ratio) | 1.0 | (0.8-1.2) |
| CRP (C-Reactive Protein) | 17 | (0-8 mg/l) |
| Sodium | 146 | (135-147 mmol/L) |
| Potassium | 3.2 | (3.3-5.0 mmol/L) |
| Chloride | 99 | (99-103 μmol/L) |
| Urea | 10.8 | (2.5-6.4 mmol/L) |
| Creatinine | 98 | (62-115 mmol/L) |
| Total protein | 66 | (60-80 g/L) |
| Albumin | 37 | (35-50 g/L) |
| Corrected calcium | 2.3 | (2.1-2.6 mmol/L) |
| Phosphate | 1.0 | (1.0-1.5 mmol/L) |
| Magnesium | 0.8 | (0.8-1.3 mmol/L) |
| Cortisol | 18 | (2.8-23 μg/dL) |
| Thyroid stimulating hormone | 11.3 | (9-30 mIU/L) |
| Free T4 | 14.2 | (10-26 pmol/L) |
| C3 complement | 0.9 | (0.5-1.53 g/L) |
| C4 complement | 0.8 | (0.2-1 g/L) |
| Cytometric analysis of peripheral blood mononuclear cells | ||
| CD3 + | 70%; 3.8 x10 9 /L | (49-77%; 1.90-5.90 x10 9 /L) |
| CD3 + CD4 + | 45%; 2.4 x10 9 /L | (31-56%; 1.4-4.3 x10 9 /L) |
| CD3 + CD8 + | 25%; 1.4 x10 9 /L | (12-24; 0.5-1.7x10 9 /L) |
| CD19 + | 21%; 1.1 x10 9 /L | (11-41; 0.43-3.0x10 9 /L) |
| NK CD3-/CD16+ | 9%; 0.5 x10 9 /L | (3-15; 0.16-0.95x10 9 /L) |
| IgE | 980 | (<15 IU/mL) |
| Antinuclear antibodies | Positive (homogeneous) | |
| Anti-pancreatic islet autoantibodies | Positive | |
| Anti-glutamic acid decarboxylase (anti-GAD65) | Positive | |
| Anti-insulin autoantibodies | Positive | |
| Direct Coombs test | Positive | |
| Anti-enterocyte KD75 autoantibodies | Positive | |
| Anti-smooth muscle autoantibodies | Positive | |
| 21-hydroxylase autoantibodies | Negative | |
EKG: normal
Upper gastrointestinal endoscopy with duodenal biopsy:
- normal visual aspect
- total villous atrophy and dense infiltrate of plasma cells and T cells
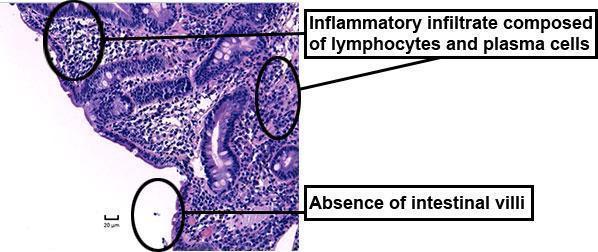
Figure 1: Duodenal biopsy
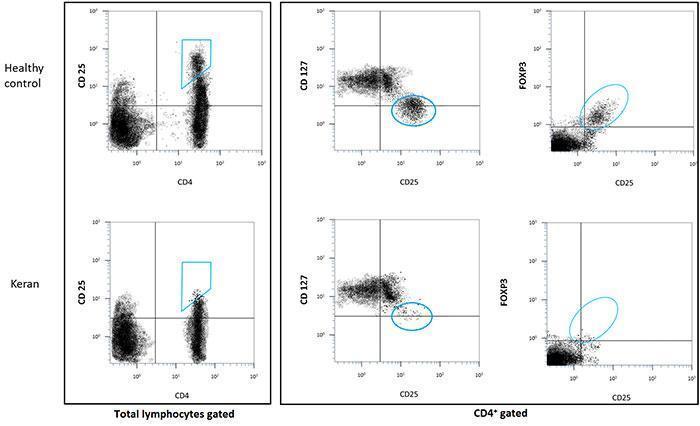
Figure 2: Analysis of CD4+ cells by flow cytometry
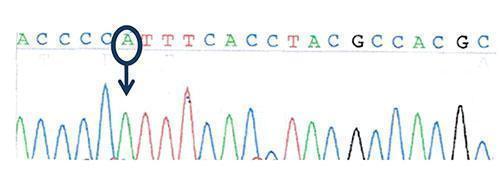
Figure 3: Sequencing of the transcription factor FOXP3 (Forkhead box protein P3) gene: hemizygous missense mutation in FOXP3 gene: c.1016 C>A, ( p.P339H)
Wild-type sequence: ACCCCCTTTCACCTACGCCACGC
Mutated sequence: ACCCCATTTCACCTACGCCACGC
Conclusion of investigations:
- normocytic anaemia with positive Coombs test: autoimmune haemolytic anaemia
- platelet and white cell count: normal
- ion and renal parameters: stigmata of dehydration with mild functional renal failure
- cortisol and thyroid: normal
- dosage of complement: normal
- immunoglobulins: hyperIgE
- anti-islet, anti-GAD65 and anti-insulin autoantibodies: autoimmune (type 1) diabetes mellitus
- villous atrophy and intestinal inflammatory infiltrate, anti-enterocyte KD75 autoantibodies, persistent watery diarrhoea: autoimmune enteropathy
- counts of CD3+CD4+ T cells, CD3+CD8+ T cells and B cells: normal
- count of CD4+/CD25hi/FOXP3+ T cells: < 0.1%
- count of CD25+/CD127low T cells: < 0.1%
- hemizygous mutation in FOXP3
Discussion
Keran was successively diagnosed with atopic dermatitis, insulin-dependent diabetes mellitus, villous atrophy (flattening and disappearance of the finger-like absorptive processes of the small intestine) and autoimmune haemolytic anaemia. Additionally, hyper-IgE and history of a deceased brother at 2-months of age from autoimmune conditions and enteropathy, are evocative of IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy X-linked).
Biological tests confirmed this diagnosis with:
- lack of regulatory T cells (Tregs, defined phenotypically as CD4+CD25hi, CD4+CD127-/lo or CD4+FOXP3+ T cells),
- a mis-sense hemizygous mutation in the FOXP3 gene on the X chromosome.
IPEX disease is a very rare X-linked poly-autoimmune syndrome due to mutations in the gene encoding for the transcription factor FOXP3, which is essential for the development and function of Treg, the pillar cells of peripheral immune tolerance. A failure in central or peripheral immune tolerance has severe consequences on homeostasis of the immune system, as illustrated by Keran’s disease.
Definition of tolerance
The immune system maintains integrity of the body by, notably, recognising pathogens and eliminating them or controlling their growth. For this, it has to distinguish dangerous organisms (typically pathogens but also abnormal or cancer cells) from innocuous antigens (typically self, alimentary or harmless environmental antigens). Indeed, the immune system faces many inoffensive foreign antigens throughout life (for eg. aerial antigens in lung, surface antigens on skin, alimentary antigens and microbiota within gut). In a way, the body itself is a universe of self-antigens. An aggressive B or T cell response against such innocuous antigens may lead to an autoimmune or allergic disease.
During their development, B and T cell precursors stochastically generate unique receptors for antigens, i.e. BCRs and TCRs, whose spectrum of recognition covers an extremely broad array of molecules. This is a fascinating aspect of the adaptive immune system which is poised to generate a considerable repertoire of receptor specificities from a limited set of genes, i.e. immunoglobulin and TCR gene loci. During this process, random molecular mechanisms generate clonal specificities directed against self-antigens among many others. It should be kept in mind that self-reactivity is a normal, physiological phenomenon: autoreactive T cells and autoantibodies are naturally present at varied levels within the immune system but are controlled by different safety checkpoints, a process referred to as ‘immunological tolerance’.
Several mechanisms participate in the prevention of undesirable responses of the adaptive immune system.
- Negative selection (or central tolerance) eliminates strongly autoreactive B and T cells during their development in the bone marrow and the thymus, respectively. Yet, this process is incomplete and self-reactive B and T cells may escape negative selection and reach the periphery of the immune system. A default in central tolerance may cause autoimmune diseases; e.g. APS-1/APECED syndrome caused by mutation in the AIRE (autoimmune regulator) gene which is important for negative selection of T cells in the thymus.
- Ignorance relates to the fact that some self-antigens are inaccessible to recognition by T cells. This is the case for instance when they are physically separated by anatomical barriers, e.g. blood-brain barrier, blood-retinal barrier etc. Nevertheless, in case of breakage of this barrier, autoimmunity may occur. One example is sympathetic ophthalmia (inflammation of the eye), a form of uveitis (inflammation of the middle layer of the eye) occurring after eye injury when ocular self-antigens are released and activate the immune system. Ignorance is not a tolerance mechanism strictly speaking because when antigens are released an immune response occurs, indicating that these T cells are not tolerant. Yet, ignorance participates in the prevention of autoimmunity.
- Peripheral tolerance prevents activation of autoreactive lymphocytes by intrinsic or extrinsic (regulation) mechanisms. As an example of intrinsic mechanism, a T cell expressing Fas (CD95) on its surface can receive signals from antigen-presenting cells expressing Fas-ligand, leading to apoptosis (peripheral deletion). Hence, a mutation of the Fas gene may cause the ALPS syndrome associating lymphoproliferation and autoimmunity. Other surface molecules including CTLA-4 and PD-1 that inhibit T cell activation also participate in peripheral tolerance. Hence, checkpoint inhibitors such as anti-PD-1 and anti-CTLA4, therapeutic monoclonal antibodies used in cancer therapy, may cause autoimmune side effects. Finally, antigen-presenting cells that are not able to deliver T cell costimulatory signals lead to T cell anergy, i.e. T cells survive but are not fully activated to fulfill their function. Extrinsic mechanisms of peripheral tolerance involve different types of regulatory cells such as Tregs or IL-10 producing Tr1 that exert a dominant negative effect on other immune cells. Among them, Tregs play the major role. They limit the responses of conventional T cells and, notably, prevent activation of autoreactive T cells.
Natural Tregs are generated by the thymus while induced Tregs differentiate from conventional T cells after activation in the periphery. Tregs were initially characterised in the mouse by their constitutive surface expression of CD25, the alpha chain of the interleukin-2 receptor. Yet, expression of CD25 is not Treg-specific since conventional T cells also transiently express CD25 after activation. In humans, Tregs were initially characterised by a higher level of CD25 expression (CD4+ CD25hi) than conventional CD4+ CD25+ activated T cells. Another phenotypic characteristic is their low level of CD127 expression (CD4+ CD25+ CD127-/lo). Importantly, they specifically express the transcription factor FOXP3 which is essential for their differentiation and function. This is an intracellular molecule whose staining necessitates cell permeabilisation to evaluate the frequency of CD4+ CD25+ FOXP3+ cells.
Tregs use many different mechanisms to regulate immunity such as T-cell deprivation of interleukin-2 (by binding to highly-expressed CD25), production of inhibitory cytokines (such as IL-10, TGF-β, IL-35) or other soluble mediators (such as adenosine), down-regulation of dendritic cell functions (such as CTLA-4 mediated blocking of dendritic cell costimulatory ligands, i.e. CD80 and CD86) or even cell killing.
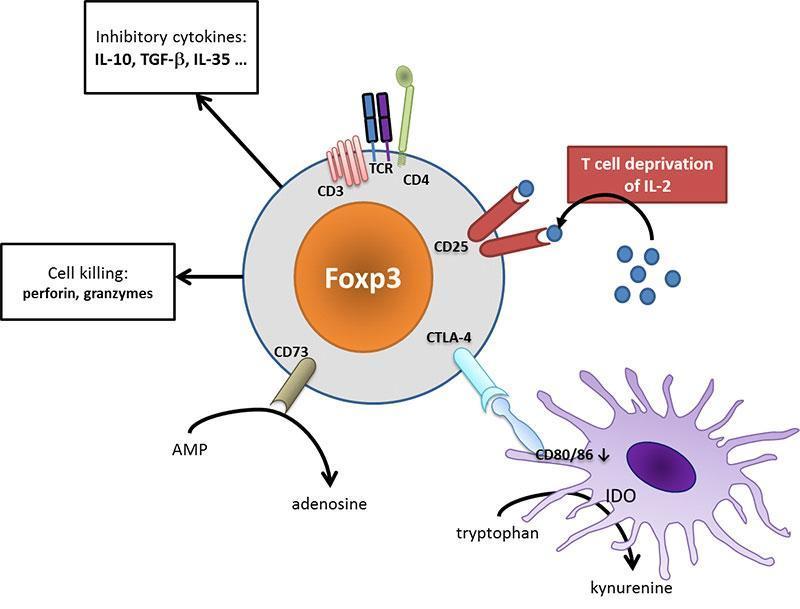
Figure 4: Tregs act at different cellular levels to regulate immunity. CD25 on Tregs efficaciously binds IL-2, depriving effector T cells from their limiting activator cytokine. Tregs may secrete different inhibitory cytokines such as IL-10 or TGF-β, leading to a tolerogenic environment. They can metabolize nucleotides to produce adenosine that acts on the inhibitory A2a receptor on immune cells. Dendritic cell are turned to a tolerogenic state (down-regulation of CD80/CD86 costimulatory ligands, production of IDO which produces inhibitory kynurenine). Ultimately, Tregs may even exert cytotoxic effect and provoke effector cell death. [Audrey Aussy, adapted from Caridade et al. 2013, PMID: 24302924]
To fight against and cope with a versatile and changing antigenic world (even ‘self’ changes with age, puberty, environment), adaptive immune systems have developed unique biological mechanisms for the generation of repertoire diversity (like somatic mutations that override the basic laws of genetics such as the conservative replication of DNA) and subsequent regulatory pathways. Keran’s clinical case illustrates the importance of peripheral tolerance since the lack of Tregs is lethal in the absence of therapy. The absence leads to unleashed activation of autoreactive responses with the contemporaneous occurrence (occurring in the same period of time) of several autoimmune diseases. The IPEX syndrome also illustrates that central deletion is not sufficient to prevent autoimmunity because Tregs are indispensable, in particular in the gut environment where most antigens are exogenous and cannot be tolerated by central mechanisms. Similarly, APS-1/APECED syndrome in which central deletion is severely impaired teaches us that Tregs alone are not sufficient to control autoreactive T cells when central tolerance is defective.
The milestones of Tregs and FOXP3 discovery
The existence of a population of suppressive cells (now known as Tregs) was suspected for a long time, before Tregs could be characterized phenotypically. For instance, when thymectomy (surgical removal of the thymus) is performed before day 3 of life, mice develop several autoimmune disorders, demonstrating that autoreactive T cells are produced at (and even before) birth. In contrast, when thymectomy is performed after day 3, mice grow normally without signs of autoimmunity, demonstrating that suppressive cells are produced from day 3 on. The latter control the activity of the former.
In 1995, Sakaguchi et al. demonstrated that transfer of CD4+CD25– T cells to an alymphocytic host provokes multiple spontaneous autoimmune diseases that can be prevented by addback of CD25+ cells. This experiment: (1) confirms that a normal repertoire (the donor has no autoimmune disease) physiologically contains autoreactive T cells; (2) demonstrates that CD4+CD25+ T cells represent a population of naturally occurring Tregs that control the activity of autoreactive T cells, and (3) suggests that Treg transfer might be used for therapy of autoimmune diseases (Sakaguchi et al., 1995).
IPEX was described in 1982 in a large family with 19 affected males across five generations, as a lethal X-linked disease (Powell et al., 1982). Then, Chatila et al. identified mutations in two unrelated patients with IPEX syndrome (Chatila et al., 2000). The IPEX syndrome was found to be the human equivalent of the Scurfy phenotype, a mouse model of lymphoproliferation (enlarged spleen and nodes) and autoimmunity. These mice have a 2-base pair insertion in a gene they named Scurfin, resulting in a truncated FOXP3 protein. At the same time, two teams independently established that FOXP3 was the human homolog of the mouse Scurfin gene encoding FOXP3 (Bennett et al., 2001; Wildin et al., 2001). Since it was not known yet that FOXP3 was specifically expressed by Tregs, authors suspected that the genetic defect caused hyperactivation of autoreactive T cells rather than a defect in regulation. To date, more than 70 mutations spanning the whole FOXP3 gene have been identified in IPEX patients.
In 2003, Sakaguchi et al. discovered that FOXP3 is selectively expressed by Tregs, and that its transduction confers regulatory phenotype and properties to conventional (non regulatory) T cells (Hori et al., 2003). The central role of FOXP3 in Tregs was subsequently confirmed by invalidation of the FOXP3 gene in mice (Fontenot et al., 2003) that recapitulated the scurfy phenotype with lymphoproliferation and autoimmunity.
The first demonstration of a role for Tregs in a classical autoimmune disease was brought by Salomon et al. who established that invalidation of the CD28 gene in autoimmune-prone NOD mice, reduced Treg numbers and accelerated type 1 diabetes (Salomon et al., 2000). Then, involvement of quantitative and/or qualitative Treg deficiencies were found in patients with many types of inflammatory or autoimmune diseases (Viglietta et al., 2004; Boyer et al., 2004; Cao et al., 2004).
In the 2010’s, Tregs started to move to therapy. Many approaches have been considered among which depletion of Tregs to augment T cell reactivity in malignant haemopathies (Maury et al., 2010), or transfer of Tregs to combat GVHD (Brunstein et al., 2011) or autoimmunity (Bluestone et al., 2015). Since low-dose IL-2 was effective in treating type 1 diabetes in NOD mice (Grinberg-Blayer et al., 2010), an alternative to cell therapy is cytokine-mediated in vivo Treg mobilisation (Saadoun et al., 2011 ; Koreth et al., 2011).
Download images for this case
Treatment
Patient’s treatment plan
Keran immediately benefited from nutritional support and insulin therapy for his autoimmune diabetes. Glucocorticoids were then quickly started in combination with cyclosporine. Atopic dermatitis, insulin-dependent diabetes mellitus, auto-immune enteropathy and autoimmune haemolytic anaemia improved in the first two months of this treatment.
Four months after initiation of immunosuppressive therapy, diarrhoea and dermatitis reappeared, while hypothyroidism was diagnosed. The indication of hematopoietic stem cells transplantation (HSCT) was then considered. One of Keran’s sisters had identical HLA haplotypes and was used as donor.
Recommended treatments and adjunct therapies
IPEX is a very rare disorder and therapeutic strategies are still based on the experience of sporadic cases reported in the literature.
The first-line of care for IPEX syndrome is immediate supportive treatment to control the different symptomatic manifestations such as parenteral nutrition for enteropathy (a disease of the intestinal tract), replacement therapy for endocrine disorders, antibiotic-prophylaxis to prevent infectious episodes, haemocomponents for cytopenias (a deficiency of a type of blood cells) or intravenous immunoglobulins for chronic diarrhoea-associated hypogammaglobulinemia (an abnormally low concentration of gamma globulin in the blood and increased risk of infection).
Monotherapy or a combination of immunosuppressive molecules only have partial efficacy, but are recommended to primarily control autoimmune disorders. Glucocorticoids (prednisone and methylprednisolone) are commonly used to limit progression of organ damage (Gambineri et al., 2008). Other immunosuppressive drugs may be combined with glucocorticoids to increase efficacy and to decrease steroid doses. Cyclosporine, tacrolimus and azathioprine are classically added to steroids. Rapamycine is another possibility.
These measures aim to control and stabilize all autoimmune disorders, but the only curative treatment is allogenic HSCT, performed as soon as possible (Baud et al., 2001). Transplantation of autologous FOXP3 gene-modified HSC may represent a perspective for the future.
Possible prevention strategies
As an X-linked monogenic disease, the only prevention strategy is to propose neonatal diagnosis to families known to harbour the mutation.
Download images for this case
Final Outcome
Outcome of patient’s treatment and the resulting physical state
After bone marrow transplantation, Keran suffered from two infectious complications, which were successfully treated. His clinical conditions improved and he returned home five months later. He is now 3 years old and healthy.
Download images for this case
References
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151-64. PMID: 7636184
Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr. 1982;100(5):731-7. PMID:7040622
Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, Bowcock AM. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest. 2000;106(12):R75-81. PMID:11120765
Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20-1. PMID:11137993
Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18-20. PMID:11137992
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003;299(5609):1057-61. PMID:12522256
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330-6. PMID:12612578
Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12(4):431-40. PMID:10795741
Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199(7):971-9. PMID:15067033
Boyer O, Saadoun D, Abriol J, Dodille M, Piette JC, Cacoub P, Klatzmann D. CD4+CD25+ regulatory T-cell deficiency in patients with hepatitis C-mixed cryoglobulinemia vasculitis. Blood. 2004;103(9):3428-30. PMID:14684420
Cao D, van Vollenhoven R, Klareskog L, Trollmo C, Malmström V. CD25brightCD4+ regulatory T cells are enriched in inflamed joints of patients with chronic rheumatic disease. Arthritis Res Ther. 2004;6(4):R335-46. PMID:15225369
Maury S, Lemoine FM, Hicheri Y, Rosenzwajg M, Badoual C, Cheraï M, Beaumont JL, Azar N, Dhedin N, Sirvent A, Buzyn A, Rubio MT, Vigouroux S, Montagne O, Bories D, Roudot-Thoraval F, Vernant JP, Cordonnier C, Klatzmann D, Cohen JL. CD4+CD25+ regulatory T cell depletion improves the graft-versus-tumor effect of donor lymphocytes after allogeneic hematopoietic stem cell transplantation. Sci Transl Med. 2010;2(41):41ra52. PMID:20650872
Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, Defor T, Levine BL, June CH, Rubinstein P, McGlave PB, Blazar BR, Wagner JE. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011;117(3):1061-70. PMID:20952687
Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, Tang Q. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7(315):315ra189. PMID:26606968
Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, Cagnard N, Carpentier W, Tang Q, Bluestone J, Chatenoud L, Klatzmann D, Salomon BL, Piaggio E. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med. 2010;207(9):1871-8. PMID:20679400
Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, Sene D, Cacoub P, Klatzmann D. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. 2011;365(22):2067-77. PMID:22129253
Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP 3rd, Armand P, Cutler C, Ho VT, Treister NS, Bienfang DC, Prasad S, Tzachanis D, Joyce RM, Avigan DE, Antin JH, Ritz J, Soiffer RJ. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365(22):2055-66. PMID:22129252
Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, Bonfanti R, Sznajer Y, Tommasini A, Lawitschka A, Junker A, Dunstheimer D, Heidemann PH, Cazzola G, Cipolli M, Friedrich W, Janic D, Azzi N, Richmond E, Vignola S, Barabino A, Chiumello G, Azzari C, Roncarolo MG, Bacchetta R. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol. 2008;122(6):1105-1112.e1. PMID:18951619
Baud O, Goulet O, Canioni D, Le Deist F, Radford I, Rieu D, Dupuis-Girod S, Cerf-Bensussan N, Cavazzana-Calvo M, Brousse N, Fischer A, Casanova JL. Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) by allogeneic bone marrow transplantation. N Engl J Med. 2001;344(23):1758-62. PMID:11396442
Download images for this case
Evaluation – Questions & answers
Cite mechanisms that participate in the prevention of undesirable responses of the adaptive immune system.
Negative selection (or central tolerance), ignorance, peripheral tolerance by intrinsic (anergy) or extrinsic (Tregs) mechanisms.
Cite different mechanisms Tregs use to exert their suppressive functions.
T cell deprivation of interleukin-2 by high CD25 expression, production of inhibitory cytokines (IL-10, TGFb, IL-35) or other molecules (adenosine), down-regulation of dendritic cell functions, cell killing.
What are the main 3 conclusions of Sakaguchi’s 1995 experiment in which transfer of CD4+CD25- T cells to an alymphocytic host provoked multiple autoimmune diseases that were prevented by CD25+ T cell addback?
(1) a normal repertoire physiologically contains autoreactive T cells, (2) CD4+CD25+ T cells are a population of naturally occurring Tregs that control the activity of autoreactive T cells, and (3) Treg transfer might be used for therapy of autoimmune diseases.
What cytokine are Tregs critically dependent on?
interleukin-2 (IL-2)
What phenotype can identify blood Tregs in patients using flow cytometry?
CD4+ CD25hi ; CD4+ CD127-/lo ; CD4+ FOXP3+
Download images for this case
Multiple Choice Questions
Earn 1 HPCSA or 0.25 SACNASP CPD Points – Online Quiz



