





In a recent study, researchers uncover how changes in joint immune cells may explain treatment resistance in rheumatoid arthritis.
Rheumatoid arthritis (RA) is a chronic autoimmune disease that causes painful inflammation and damage in the joints and for many patients, even the best medications eventually stop working. Now, researchers have uncovered a key reason why: as the disease progresses, immune cells inside the joints change their identity, becoming resistant to common anti-inflammatory therapies (Figure 1).
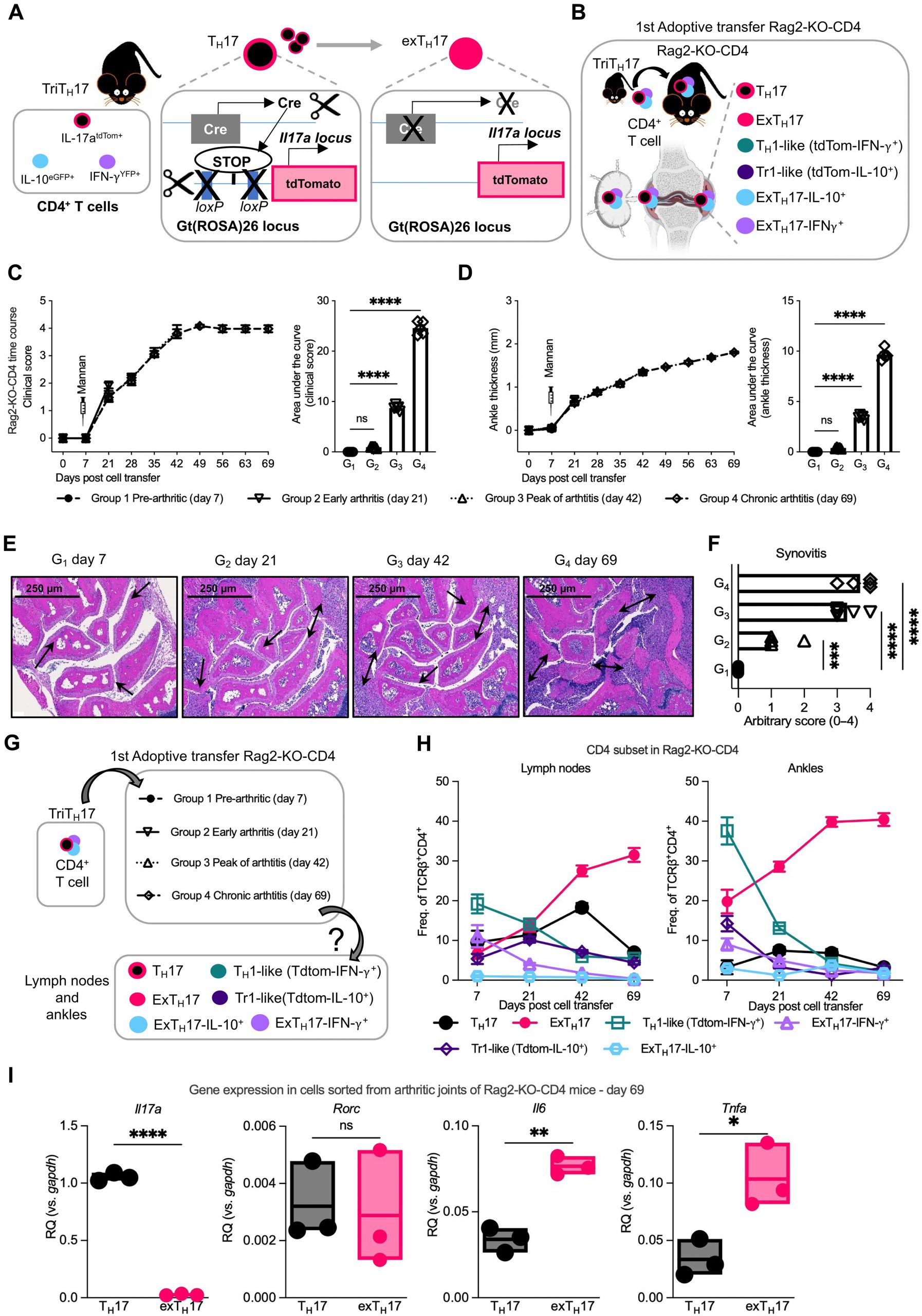
Figure 1: TH17 cells convert into exTH17 cells and become a prominent T cell population in joints and lymph nodes of SKG mice with chronic arthritis. (A) Scheme of triple TH17 cell fate mapping SKG mice (triTH17 cell: B6.Il17aCre+/−.Il10eGFP/eGFP.IfngYFP/YFP.R26tdTomfl/fl.Zap70SKG/SKG.H2d/d) used as a source of cells for the experimental arthritis model. (B) Scheme of arthritis model. TriTH17 CD4+ T cells (1 × 106) were adoptively transferred into B6.Rag2−/−.H2d/d (Rag2-KO), and arthritis severity and T cell fate in joint and lymph nodes were assessed. (C and D) Time course of arthritis includes four time points (one group of mice per time point) with N = 5 mice for each group. (C) Arthritis assessment, evaluated by clinical score (left) with area under the curve (AUC, right). Boxes under the figure list time points used in (E) to (H). Ankle thickness measurement (left) and AUC (right). Ankle thickness was plotted as the difference between measured thickness and baseline thickness for that time point. Statistics were calculated on the AUC. (E) Representative histology for Rag2-KO-CD4 mice assessed at the indicated time points, H&E staining. Arrows indicate areas of inflammation. (F) Histological quantification of synovitis at the indicated time points. (G) End points for each group in time course of arthritis experiment (N = 5 mice per point). (H) Frequency (% of TCRβ+CD4+) of T cell subpopulations [TH17, exTH17, Tr1-like (Tdtom−IL10+), TH1-like (Tdtom− IFN-γ+), ExTH17-IL10+, and ExTH17–IFN-γ+ cells] in lymph nodes (left) and joints (right) of Rag2-KO-CD4 from day 7 after CD4+ T cell injection to day 69 of arthritis. (I) mRNA expression of indicated genes in sorted TH17 cells versus exTH17 cells from arthritic ankles of Rag2-KO-CD4 (N = 3). Graphs show relative quantification (RQ) versus housekeeping gene (Gapdh). Graphs in (C), (D), and (H) show grouped summary data plotted at mean ± SEM, each line corresponding to a different group of mice, and each point in histograms represents data from an individual mouse [statistics for (H) are shown in fig. S2C]. Histograms in (F), (H), and (I) show mean ± SEM, and each point in histograms represents data from an individual mouse. [(C), (D), and (F)] One-way ANOVA with Dunnett’s multiple comparisons test, reference group is G1; (I) unpaired t test; two tailed. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant.
Many autoimmune diseases, including some forms of arthritis, can be effectively treated by blocking interleukin-17 (IL-17), an inflammatory protein that fuels immune attacks. But IL-17–targeted therapies have consistently underperformed in established rheumatoid arthritis, leaving scientists puzzled.
By examining human joint tissue samples and mouse models, the team found that, over time, immune cells in rheumatoid joints stop producing IL-17 altogether. Yet these cells don’t become less inflammatory, instead, they reprogram themselves to sustain inflammation through alternative, IL-17–independent pathways.
The researchers also identified synoviocytes, nonimmune cells that line the joint and secrete lubricating synovial fluid as powerful influencers of this immune transformation. Over time, these synoviocytes appear to reshape immune cell behaviour, pushing them toward a chronic inflammatory state that is resistant to standard immunosuppressive treatments.
Advanced spatial biology techniques allowed the team to map how these cellular interactions unfold within the complex joint environment.
The findings suggest that early intervention may be crucial, before immune cells become permanently reprogrammed. In the future, therapies targeting the cellular crosstalk between immune cells and synoviocytes may help reverse or prevent this transition, paving the way for more durable, precision-based treatments.
Rheumatoid arthritis treatment resistance may stem not from failed drugs, but from evolving immune cells shaped by the joint’s own tissue environment. By uncovering how these cells rewire themselves, scientists are laying the groundwork for more effective, personalized therapies to stop arthritis at its roots.
Journal article: Zoccheddu, M., et al. 2025. TH17 cells converted into exTH17 cells sustain rheumatoid-like IL-17–independent inflammatory arthritis. Science Immunology.
Summary by Stefan Botha



