





Cancer immunotherapy has a recurring problem: tumors evolve. A therapy that eliminates one cancer population can leave behind a resistant clone that lacks the targeted antigen. That evolutionary escape is a major reason for relapse after single-antigen CAR T cell therapy. A recent study in Cell Reports Medicine reports an engineering strategy designed to shrink those escape routes by giving each T cell three independent ways to recognize and kill tumor cells (Figure 1).
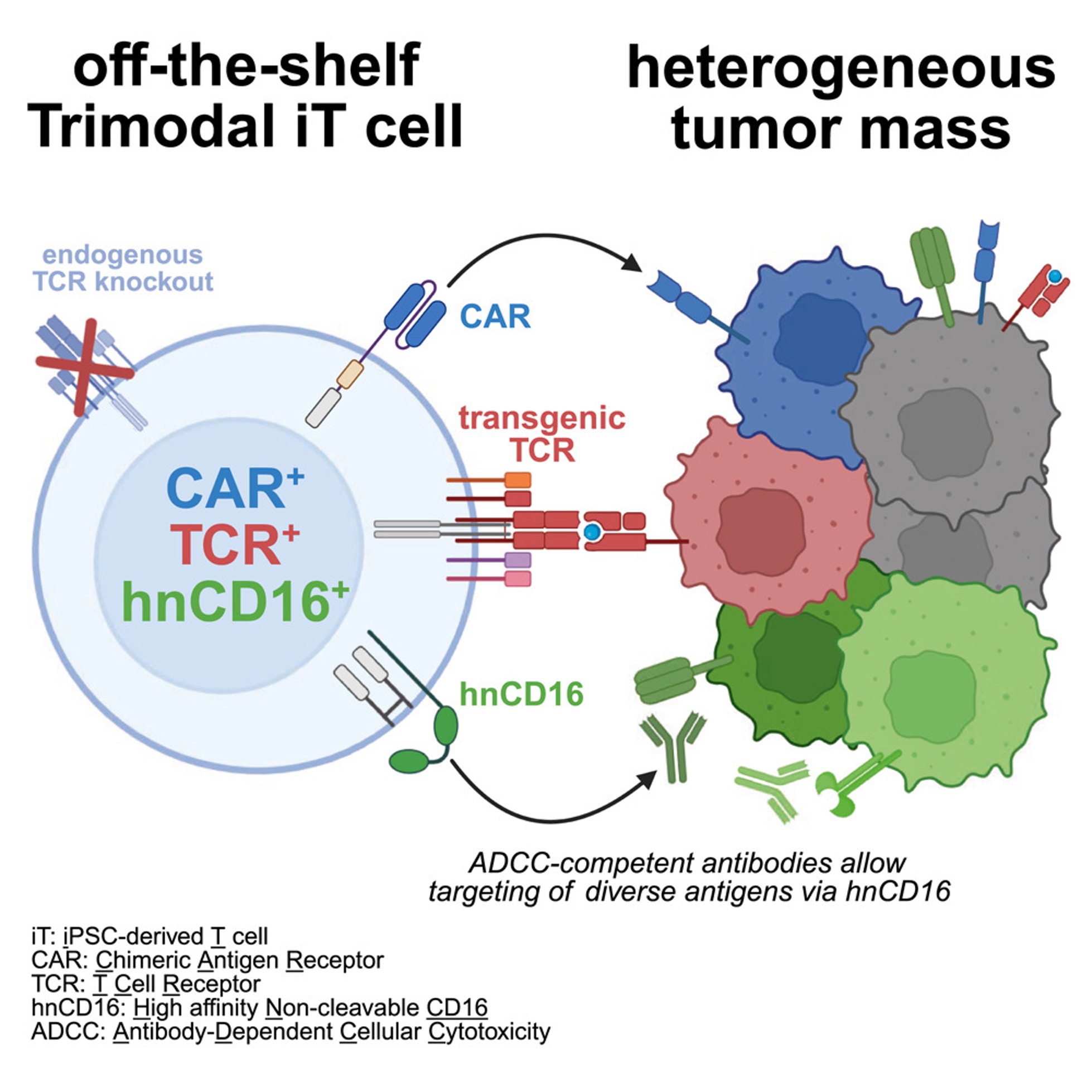
Figure 1: Graphical abstract.
The authors created “trimodal” T cells derived from induced pluripotent stem cells (iPSCs). Each engineered cell carries: (1) a chimeric antigen receptor (CAR) targeting a surface antigen, (2) a transgenic T cell receptor (TCR) recognizing a peptide presented on MHC, and (3) a high-affinity, non-cleavable CD16 receptor (hnCD16) that enables antibody-dependent cellular cytotoxicity (ADCC). In practical terms, this means a single immune cell can kill through three orthogonal targeting modes, rather than relying on one receptor and hoping the tumor does not escape.
The logic is simple and strong. CARs recognize extracellular antigens, but tumors can downregulate or lose these targets. TCRs recognize intracellular proteins through peptide–MHC presentation, which expands the antigen space beyond the cell surface. CD16 binds the Fc portion of antibodies. If a tumor cell is coated with a therapeutic antibody, a CD16-positive effector can bind and kill it. By equipping T cells with hnCD16, the authors make the same T cell product redirectable using existing monoclonal antibodies, potentially allowing clinicians to adjust targeting without re-engineering the cell therapy.
To build this system, the team used CRISPR-mediated knock-in to place a cassette encoding the CAR and hnCD16 into both alleles of the TRAC locus in iPSCs. Targeting TRAC helps enforce uniform expression and disrupts endogenous TCR activity. They then introduced an antigen-specific TCR via lentiviral transduction. The engineered iPSCs were differentiated into T cells, yielding a highly uniform population. By the end of differentiation, more than 98% of cells co-expressed the CAR, the transgenic TCR, and hnCD16, and global expression profiling showed these cells clustered with CD8 T cells.
A key concern with adding CD16 to T cells is whether it actually triggers functional T cell signaling. The authors addressed this directly. Crosslinking hnCD16 induced rapid phosphorylation of CD3ζ and downstream TCR signaling proteins including ZAP70 and ERK. This confirmed that hnCD16 engagement can drive canonical activation pathways. Functionally, the engineered cells performed ADCC in vitro. They killed target cells efficiently only when the correct therapeutic antibody was present, supporting specificity rather than nonspecific activation.
The central claim of the paper is not just that each module works alone, but that combining them prevents escape in heterogeneous tumors. The authors designed “stress test” experiments using mixed tumor populations where each subclone could be eliminated by only one of the three targeting mechanisms. When T cells lacked one modality, the corresponding tumor subpopulation escaped and expanded. When all three modalities were present, residual tumor burden was lowest and cytokine production was highest, consistent with sustained engagement across the entire mixed population.
They then moved to in vivo xenograft models using immunodeficient mice bearing mixtures of leukemia subclones with different antigen profiles. Dual-modality approaches improved control relative to single-modality conditions, but the trimodal strategy provided the most consistent reduction in tumor burden and improved survival, especially when the ADCC arm was paired with the relevant antibody. The in vivo data support the same principle seen in vitro: parallel targeting reduces the evolutionary space for antigen escape.
The study is also clear about limitations. Persistence remains a bottleneck. Although trimodal T cells were detectable weeks after infusion, their numbers did not expand enough to prevent late tumor outgrowth in some settings. In addition, several experiments rely on engineered tumor lines with defined antigen patterns, which improves mechanistic clarity but may not fully capture the complexity of human tumors. Sample sizes in some in vivo readouts are modest, so effect sizes should be interpreted carefully.
Even with these caveats, the paper makes a strong conceptual point. Multi-modality within a single T cell can provide robust coverage against heterogeneity and antigen loss. The iPSC platform adds another layer of appeal by enabling uniform, clonally engineered, potentially off-the-shelf products. Future work will likely focus on extending persistence and translating this architecture into more physiologic tumor models.
Journal article: Yang BH, et al. 2025. iPSC-derived trimodal T cells engineered with CAR, TCR, and hnCD16 modalities can overcome antigen escape in heterogeneous tumors. Cell Rep Med.
Summary by Gaurang Telang



