





A new study has provided one of the most detailed views yet of how lung disease begins in children with cystic fibrosis (CF), revealing that immune dysfunction and tissue damage are already well established during the preschool years (Figure 1). The work challenges the assumption that current breakthrough CF therapies are sufficient to fully protect the developing lung from irreversible injury.
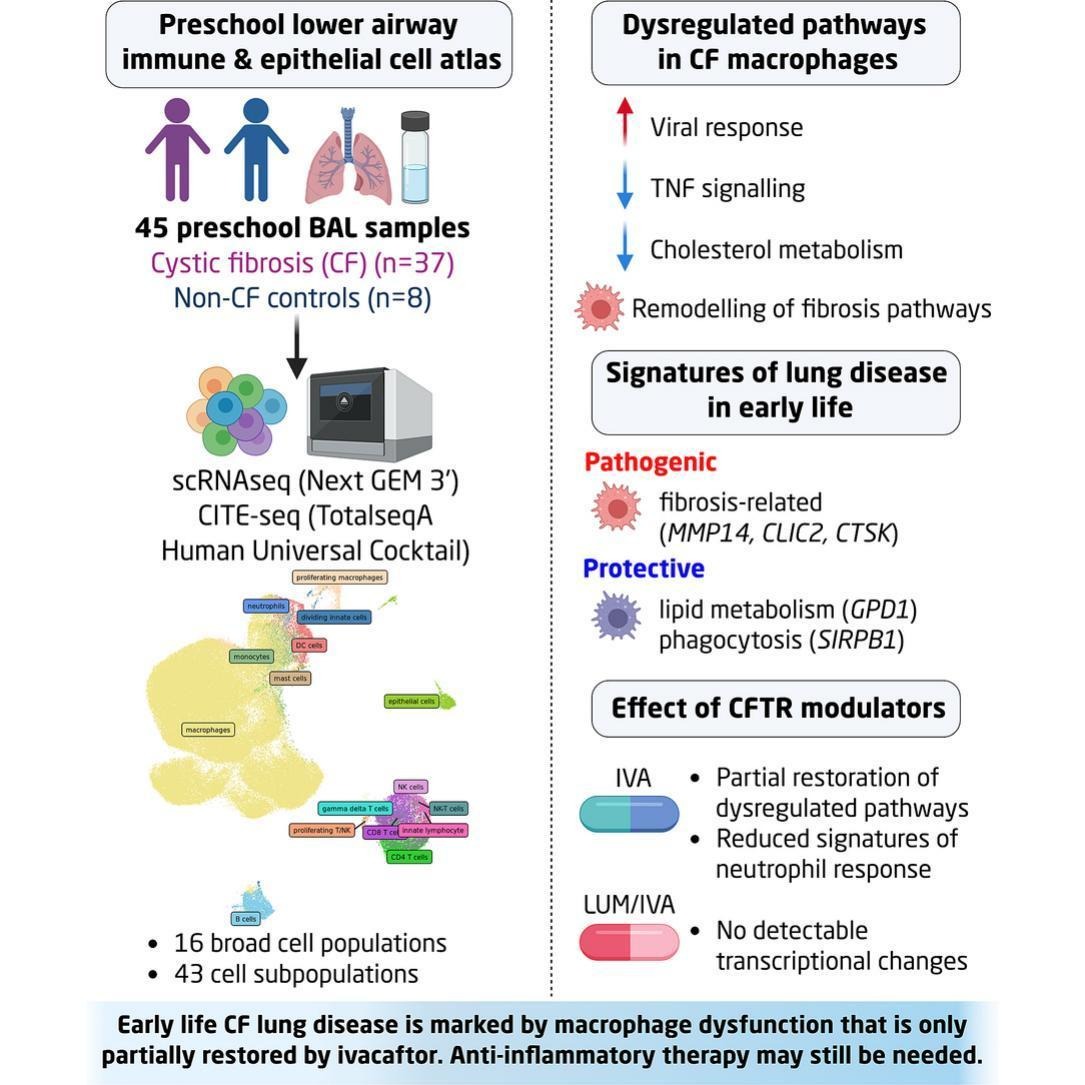
Figure 1: Graphical abstract.
Using single-cell RNA sequencing and protein profiling, the researchers generated the largest lower-airway cellular atlas to date for early-life cystic fibrosis. The study analysed more than 190,000 cells from bronchoalveolar lavage samples collected from 37 children aged between five months and six years. Across these samples, investigators identified 43 distinct immune and epithelial cell populations, providing an unprecedented map of the cellular landscape of the young CF lung.
A major finding was that immune abnormalities emerge remarkably early in life. Macrophages, the key innate immune cells responsible for clearing pathogens and coordinating inflammatory responses, showed widespread dysfunction in preschool-aged children with CF. These macrophages displayed altered inflammatory signalling, disrupted cholesterol metabolism, and activation of profibrotic pathways associated with tissue scarring and bronchiectasis. Importantly, these abnormalities were even more pronounced in children who had already begun developing bronchiectasis, an irreversible structural lung disease that significantly contributes to long-term respiratory decline in cystic fibrosis.
The study also examined the effects of CFTR modulator therapies such as ivacaftor, which target the underlying genetic defect in cystic fibrosis and have transformed patient outcomes in recent years. While these therapies altered certain inflammatory programs, the data suggest that they do not fully normalize immune dysfunction within the lungs of young children. This raises important questions about whether current treatments alone are sufficient to prevent progressive airway remodelling and chronic lung injury.
The findings reinforce the growing idea that cystic fibrosis is not only a disorder of defective ion transport and mucus accumulation, but also a disease of persistent immune dysregulation. Thickened mucus creates an environment that promotes chronic infection, but impaired macrophage function may further compromise the lung’s ability to effectively clear pathogens and resolve inflammation, potentially accelerating tissue damage from a very early age.
Importantly, the work identifies a potential therapeutic window during early childhood where intervention may still prevent irreversible structural lung disease. The authors suggest that combining CFTR modulators with targeted anti-inflammatory therapies could provide greater protection against long-term lung injury. This approach may be particularly important given that bronchiectasis can develop silently during the preschool years, often before substantial clinical symptoms become apparent.
Journal article: Maksimovic, J., et al. 2026. Single-cell profiling of BAL in preschool cystic fibrosis reveals macrophage dysregulation and ivacaftor-modified inflammatory programs in the early life lung. Mucosal Immunology.
Summary by Stefan Botha



