





A new study uncovers how immune cells can exploit aberrant protein synthesis in cancer cells to identify shared, highly expressed targets for adoptive T-cell therapy (Figure 1).
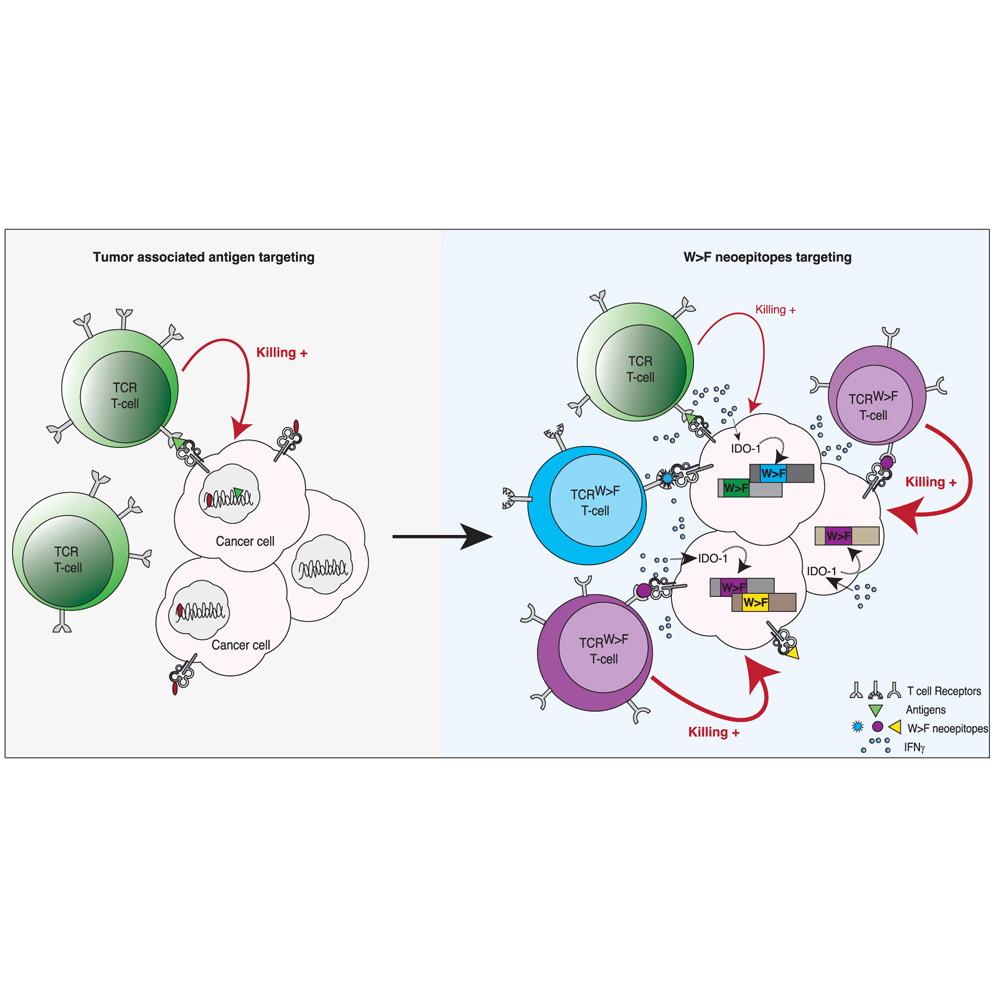
Figure 1: Graphical abstract.
When cancer cells are exposed to prolonged Interferon‑gamma (IFNγ), they upregulate the enzyme Indoleamine 2,3‑dioxygenase 1 (IDO1), which depletes cellular tryptophan. This shortage of tryptophan triggers a form of ribosomal frameshifting in which the tryptophan (W) codon is reassigned to phenylalanine (F), creating W→F substitutions in proteins. The study shows that these aberrant peptides become processed and presented on HLA molecules, turning translational “mistakes” into neoepitopes the immune system can recognize.
Using immunopeptidomic analyses (i.e., profiling the peptides presented by HLA molecules), the research team found hundreds of W→F neoepitopes. Most of these were presented by the allele HLA‑A24:02. Eur.nl+1 They identified in particular one epitope derived from the gene TMBIM6 with a W→F substitution (“TMBIM6W>F”) that had the broadest expression across many cancer cell lines. A specific T-cell receptor (TCR) named TCRTMBIM6W>F.1 was shown to bind this epitope in complex with HLA-A24:02 with high affinity and specificity. These T cells were activated by cancer cells under tryptophan deprivation, but not by non-cancer cells, indicating a good therapeutic window.
In vivo experiments provided proof of concept: the TCRTMBIM6W>F.1 T cells could mediate tumour killing. Interestingly, these were used in combination with another TCR (TCRMART1) to amplify generation of W→F neoepitopes in tumour cells, thereby enhancing the effect. This provides a blueprint for adoptive T-cell therapy that targets these inducible, shared neoepitopes rather than fully private mutations.
Why this matters:
- Shared targets: Because these W→F neoepitopes arise from a common mechanism (tryptophan starvation → frameshift → W>F), they may be present across many cancer types, unlike purely patient-specific mutation‐derived neoepitopes.
- High expression & inducible: The epitopes are broadly expressed in cancer cells (especially under IFNγ/IDO1 conditions) and are not (or little) in normal cells, offering specificity.
- New class of neoepitopes: This expands the universe of targetable neoantigens beyond DNA-level mutations to include translational/frameshift errors induced by metabolic stress.
- Clinical potential: Adoptive T-cell therapies (engineered TCRs) directed at these epitopes might overcome some limitations of current neoantigen-based therapies (which often require personalised production, and the targets vary widely between patients).
Looking ahead
The authors propose that TCRs like TCRTMBIM6W>F.1 could serve as “off-the-shelf” tools once HLA compatibility is managed (here HLA-A*24:02). Major next steps include:
- Assessing safety and off-target risks in humans.
- Evaluating which cancers and patient subpopulations express the W→F neoepitope sufficiently.
- Investigating how tumour microenvironmental conditions (e.g., IFNγ exposure, IDO1 levels, tryptophan scarcity) shape presentation of W→F neoepitopes in vivo.
- Combining this approach with standard immunotherapies (checkpoint inhibitors) or metabolic modulators to maximise epitope generation and T-cell effect.
This work highlights a novel mechanism of neoepitope formation, tryptophan starvation leading to W→F codon reassignment, and leverages it for adoptive T-cell therapy. Shared, inducible neoepitopes like TMBIM6W>F may open up new paths to treat cancers with TCR therapies, sidestepping some of the hurdles of personalised neoantigen approaches.
Journal article: Champagne, J., et al. 2025. Adoptive T cell therapy targeting an inducible and broadly shared product of aberrant mRNA translation. Immunity.
Summary by Stefan Botha



